Новые данные о роли α-синуклеина в патогенезе болезни Паркинсона
Белки — основа жизненных процессов. Неправильная пространственная укладка белков является наиболее серьезной угрозой для функционирования и выживания клеток. Известно, что не более 70% из вновь синтезированных белков имеют правильную трехмерную структуру, необходимую для реализации их биологической активности. Процессы старения, различные стрессы и повреждения, связанные с патологией, существенно увеличивают количество аномальных белков, опасных для клетки. В последние годы важное значение в патогенезе болезни Паркинсона (БП) отводится нарушению конформации белка α-синуклеина, ведущему к фатальному накоплению его токсичных олигомеров в клетках, в том числе в дофамин ДА-ергических нейронах компактной части черной субстанции и дегенерации нейронов; подобный механизм играет существенную роль в патогенезе спорадической и наследственной форм БП.
Строение и физиологические функции α-синуклеина
Интерес к свойствам небольшого растворимого белка α-синуклеина резко возрос после того, как была установлена взаимосвязь между нарушением его функции и развитием ряда нейродегенеративных заболеваний, включая БП. Многолетний патологический процесс при БП характеризуется прогрессирующей потерей ДА-ергических нейронов в компактной части черной субстанции и ряде других структур мозга и образованием сферических эозинофильных цитоплазматических включений (телец Леви) в соме нервных клеток, а также веретено- или нитеподобных ветвящихся нейритов Леви и нейроаксональных сфероидов в отростках нейронов; эти включения являются морфологическим субстратом нейродегенерации. Накопление телец и нейритов Леви в нервной ткани рассматривается в качестве критического диагностического фактора БП.
Главным компонентом телец и нейритов Леви являются агрегации неправильно свернутого и токсичного для клетки высококонсервативного белка α-синуклеина с молекулярной массой 19 кДа (140 аминокислот), который локализуется в основном в аксонах и их пресинаптических терминалях. Ген α-синуклеина (SNCA) расположен на длинном плече четвертой хромосомы и состоит из семи экзонов, пять из которых транскрибируются. Агрегации α-синуклеина характерны для целой группы заболеваний, которые рассматриваются как синуклеинопатии (табл.)

Альфа-синуклеин широко распространен в мозге. В полном транскрипте, α-синуклеине-140, выделяют три области (рис. 1). N-концевой участок (основания 1-65) включает сайты по крайней мере двух аутосомно-доминантных наследственных мутаций, вызывающих БП, — замена аланина на треонин в позиции 53 (А53Т) и замена аланина на пролин в позиции 30 (А30Р); он содержит шесть неполных повторов 11-аминокислотных остатков с высококонсервативным гексамерным мотивом (KTKEGV). Предполагается, что N-концевой участок способен образовывать структуру из двух α-спиралей, соединенных коротким прямолинейным участком, и типичную для липид-связывающих доменов аполипопротеинов класса А2. Было показано, что липидное окружение стимулирует сворачивание α-синуклеина, а также ускоряет его агрегацию. Центральный гидрофобный участок (основания 66-95) включает NAC-последовательность (non-amyloid component), склонную к агрегации. Впервые NAC-пептид был выделен из амилоидных бляшек коры головного мозга пациентов, страдавших болезнью Альцгеймера. И лишь затем было обнаружено, что α-синуклеин является основным компонентом телец Леви при БП. С-концевой участок (основания 96-140) отрицательно заряжен за счет кислых аминокислот. В этом регионе локализованы несколько сайтов фосфорилирования (Туr-125, -133 и -136 и Ser-129), а также домен, отвечающий за шаперонную активность α-синуклеина (основания 125-140).
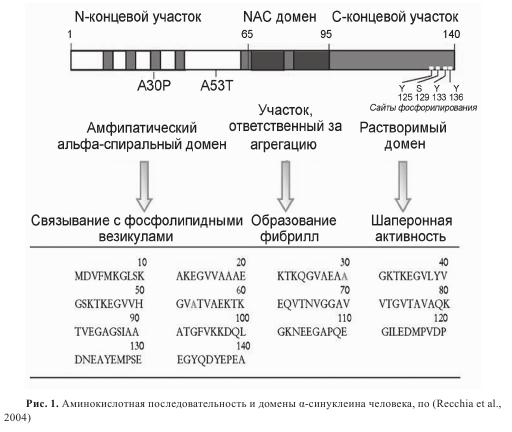
Функции этого распространенного белка, количество которого в растворимой цитозольной мозговой фракции составляет не менее 1% от всего белка, остаются недостаточно выясненными. Однако его локализация в пресинаптических терминалях, ассоциация с дистальным резервным пулом синаптических везикул и нарушение синаптической передачи при нокаутировании животных по α-синуклеину или при избыточной экспрессии этого белка позволяют считать, что α-синуклеин играет определенную роль в регуляции высвобождения нейромедиаторов, синаптической функции и пластичности.
Анализ мышей-нокаутов по гену α-синуклеина показал, что у них заметно сокращается время, необходимое для восстановления способности к выбросу ДА в стриатуме после повторной электрической стимуляции, падает уровень ДА в стриатуме и наблюдается снижение локомоторного ответа на введение амфетамина (в результате уменьшения выброса ДА в синаптическую щель и инвертирования действия дофаминового транспортера на локомоторную активность). У мышей-нокаутов по гену Snca обнаружены нарушения гиппокампальных синаптических реакций в ответ на длительную высокочастотную стимуляцию, способную истощить готовые к выбросу и резервные пулы синаптических везикул, а также нарушения процесса пополнения готовых к выбросу пулов из резервных депо. Эти результаты могут означать, что α-синуклеин контролирует пополнение синаптических везикул и их перемещение из резервного пула к области высвобождения синаптических везикул. Кроме того, показано, что истощение α-синуклеина с помощью антисмысловых олигонуклеотидов вызывает уменьшение резервного пула синаптических везикул в первичной культуре нейронов гиппокампа.
У трансгенных мышей с гиперэкспрессией человеческого α-синуклеина определены нарушения экзоцитоза синаптических везикул и уменьшение высвобождения нейромедиатора. Подобные эффекты наблюдались при гиперэкспрессии α-синуклеина в генетических моделях БП на грызунах и на клеточной линии РС12. Гиперэкспрессия α-синуклена вызывает снижение количества готовых к высвобождению везикул, рециркуляции синаптических пузырьков после эндоцитоза, размера синаптического пула рециркулирующих везикул, обратного захвата ДА в ДА-ергических терминалях.
Возможная роль α-синуклеина в регуляции синаптического гомеостаза не связана исключительно с его непосредственным взаимодействием с синаптическими пузырьками. Показано, что α-синуклеин может действовать как белок-шаперон по отношению к пресинаптическому белковому комплексу SNARE, непосредственно вовлеченному в процесс высвобождения нейромедиаторов, включая ДА. Совокупность данных свидетельствует, что α-синуклеин играет важную роль в перемещении синаптических везикул и регуляции везикулярного экзоцитоза и, возможно, контролирует ряд белков, связанных с синаптическим гомеостазом.
Тот факт, что нокауты по тому или иному синуклеину (α, β и γ) жизнеспособны, позволяет считать, что синуклеины не являются ключевыми участниками в механизмах высвобождения нейромедиатров, но, возможно, вносят свой вклад в долгосрочную регуляцию и поддержание функций нервных терминалей.
Секреция α-синуклеина и пути распространения синуклеиновой патологии
Отсутствие секреторной сигнальной пептидной последовательности в молекуле α-синуклеина привело к предположению, что он является исключительно внутриклеточным белком, и его патологическая функция долго рассматривалась в контексте внутриклеточного окружения. Однако это представление изменилось, когда было обнаружено присутствие α-синуклеина в биологических жидкостях, таких как цереброспинальная жидкость и плазма крови как у лиц с БП, так и у здоровых людей. Сделан акцент на изучении механизма высвобождения и возможные функции внеклеточного α-синуклеина.
Показано, что α-синуклеин секретируется в питательную среду из клеточных линий сего гиперэкспрессией. Определена секреция мономерных и агрегированных форм эндогенного α-синуклеина из эмбриональных нейронов коры мозга крысы.
При гиперэкспрессии в клетках нейробластомы человека SH-SY5Y α-синуклеин локализуется в больших плотных везикулах в ядре клетки и эта внутривезикулярная фракция более склонна к агрегации по сравнению с цитозольным α-синуклеином. Обнаружено что ДА увеличивает уровень секреции α-синуклеина и вызывает накопление его в везикулах. Предполагается, что высвобождение α-синуклеина усиливается в условиях клеточного стресса, способствующего накоплению неправильно уложенных и поврежденных белков.
В последние годы повысился интерес к внеклеточному α-синуклеину в связи с его потенциальной ролью в инициации и развитии синуклеинопатий. Основанием послужили работы, описывающие пошаговое, постадийное распространение патологии телец Леви при БП и присутствие α-синуклеинположительных включений наподобие телец Леви после давно проведенной мезенцефалической трансплантации пациентам с БП, свидетельствующие о распространении α-синуклеиновой патологии от клеток хозяина в трансплантированные клетки. Эксперименты, проведенные in vitro, подтвердили возможность межнейрональной передачи α-синуклеина (рис. 2). Такая передача α-синуклеина наблюдалась и в экспериментах in vivo, проведенных на трансгенных мышах, экспрессирующих α-синуклеин. Процесс передачи α-синуклеина связан, по-видимому, с захватом α-синуклеина нейронами-реципиентами посредством эндоцитоза.
Эти результаты позволяют предполагать, что α-синуклеин способен не только секретироваться клетками, но и передаваться от одних нейронов к другим; секреция α-синуклеина усиливается в условиях клеточного стресса.
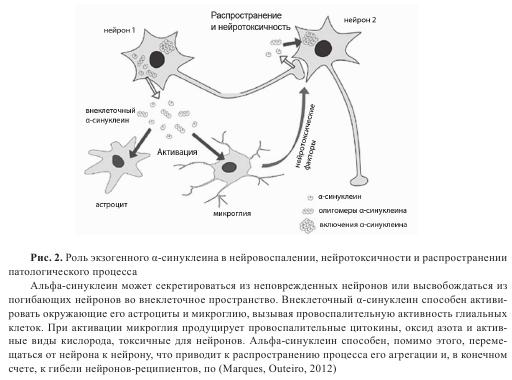
Роль α-синуклеина в нейровоспалении, нейротоксичности и клеточной гибели
Феномены секреции и межклеточной передачи α-синуклеина сами по себе не являются достаточным объяснением распространения α-синуклеиновой патологии и прогрессии нейродегенерации, типичных для БП. Существуют дополнительные данные о том, что α-синуклеин, подобно прионам, может выступать в качестве агента, катализирующего агрегацию, и способен оказывать влияние на жизнеспособность нейронов-реципиентов. Экзогенные фибриллы α-синуклеина могут вызывать формирование внутриклеточных включений, подобных тельцам Леви, в культивируемых клетках. Получены также данные о формировании включений вследствие межклеточной передачи α-синуклеина как при совместной культивации клеток, так и в экспериментах in vivo при их трансплантации (см. рис. 2). Важно подчеркнуть, что помимо стимуляции агрегации внеклеточный α-синуклеин, выделенный из нейронов, вызывает процесс апоптоза в нейронах-реципиентах.
В ряде работ отмечен нейротоксический эффект внеклеточных и секретированных олигомерных форм α-синуклеина. Олигомеры рекомбинантного α-синуклеина захватываются нейронами в культуре и запускают клеточную гибель; секретированные нейронами олигомеры оказывают токсическое действие на нейроны-реципиенты. Добавление в культуральную среду веществ, препятствующих олигомеризации, защищает нейроны-реципиенты. Следовательно, внеклеточный α-синуклеин участвует в распространении процесса нейродегенерации при БП.
Внеклеточный α-синуклеин способен запускать нейровоспалительные реакции в клетках глии. Показано, что воспалительный процесс в черной субстанции характеризуется секрецией провоспалительных и нейротоксических факторов, инициирующих дегенерацию нейронов. В цереброспинальной жидкости или сыворотке крови пациентов с БП обнаружены повышенные уровни провоспалительных цитокинов, таких как интерлейкин 1 β (IL1-β), интерлейкин 2 (IL-2), интерлейкин 6 (IL-6) или фактор некроза опухоли альфа (TNF-α).
В опытах in vitro установлено, что под влиянием α-синуклеина клетки микроглии увеличивают продукцию NO, TNF-α и IL1-β, толл-подобных рецепторов и активных форм кислорода, что приводит к усилению нейротоксичности и нейродегенерации ДА-ергических нейронов; описана прямая активация воспалительной реакции в микроглиальной клеточной линии внеклеточным α-синуклеином, полученным из нейронов.
Введение человеческого α-синуклеина дикого типа в черную субстанцию мышей вызывает экспрессию провоспалительных цитокинов и маркеров воспаления в эндотелии и активацию микроглии; при наличии внеклеточного α-синуклеина микроглия становится более восприимчивой к системным провоспалительным сигналам.
Воздействия α-синуклеина на микроглию сопровождаются также реакцией со стороны астроцитов. Передача α-синуклеина от нейронов к астроцитам показана в опытах с использованием клеточных культур и in vivo в мозге трансгенных мышей, экспрессирующих человеческий α-синуклеин. Она приводит к накоплению α-синуклеина внутри астроцитов и вызывает в них воспалительную реакцию.
Приведенные данные свидетельствуют об участии внеклеточного α-синуклеина в модуляции глиальной функции и нейровоспалении и фокусируют внимание исследователей на выяснении механизмов, за счет которых происходит межклеточная передача α-синуклеина.
Конформационные свойства α-синуклеина
Ключевой вопрос, касающийся молекулярных основ патогенеза БП и других синуклеинопатий, — какая из широкого перечня стадий агрегации α-синуклеина является наиболее токсичной для клеток. Этот перечень возможных конформаций α-синуклеина включает: а) развернутое состояние (при физиологических условиях in vivo), б) глобулярную форму (при высокой температуре, в присутствии ионов металлов, солей, некоторых гербицидов, пестицидов и при других условиях), в) α-спиральную мембрано-связанную форму (при взаимодействии α-синуклеина с синтетическими везикулами, содержащими кислые фосфолипиды), г) состояние в виде α-спиралей и β-слоев (при добавлении спиртов), д) димеры (при пролонгированной инкубации при высокой температуре), е) олигомеры, ж) крупные нерастворимые включения двух типов: аморфные агрегаты и фибриллы. Конечным этапом кластеризации α-синуклеина является образование телец Леви. Показано, что инкубация сферических олигомеров α-синуклеина с молекулярным весом 2,5-4,2 нм с мембранами, выделенными из мозга, приводит к формированию пороподобных кольцевых протофибрилл; предполагается, что именно эта стадия α-синуклеиновой агрегации приводит к гибели клетки. Тельца Леви, видимо, выполняют защитную функцию в клетке, аккумулируя токсичные для нейронов белки и предотвращая их распространение. В пользу последнего предположения свидетельствует тот факт, что при мутации белка паркина развивается аутосомно-рецессивный ювенильный паркинсонизм, не сопровождающийся образованием телец Леви, но приводящий к накоплению растворимых олигомерных форм α-синуклеина и к гибели клеток.
Ранее полагали, что молекула α-синуклеина обладает необычными конформационными свойствами, представляя собой нативно неуложенный белок, который обладает слабоупорядоченной или недоупорядоченной структурой при физиологических условиях in vivo, т.е. у него отсутствует четко заданная пространственная структура.
Работы последних лет показали, что эндогенный внутриклеточный α-синуклеин существует преимущественно в виде α-спирального уложенного тетрамера с молекулярной массой ~58 кДа. Тетрамеры α-синуклеина не образуют агрегатов, однако в случае нарушения процесса формирования тетрамеров свободные спиральные мономеры белка начинают слипаться друг с другом, образуя амилоидные волокна, которые впоследствии формируют тельца Леви в нервных клетках. Пока последовательность образования амилоидных волокон не ясна, возможно, их формируют мономеры с изначально нарушенной структурой, не способные к объединению в тетрамеры, либо функциональные тетрамеры в клетке по какой-то причине разрушаются, и высвободившиеся мономеры начинают слипаться. В любом случае разработка метода стабилизации тетрамеров α-синуклеина может стать эффективным инструментом замедления нейродегенеративного процесса, а быть может, и предотвращения развития БП.
Альфа-синуклеин и современная классификация доклинических и клинических стадий болезни Паркинсона
Основной недостаток известной классификации состоит в том, что продолжительный (до 10-30 лет) и наиболее важный для лечения период болезни, который предшествует моторным симптомам, в ней не представлен. Постепенно накапливались данные о распределении в мозге агрегаций α-синуклеина в виде телец и нейритов Леви, которые служат основанием для выделения последовательных стадий БП. При этом отмечено очень важное обстоятельство — локализация агрегаций α-синуклеина может быть связана с особенностями архитектуры нервных клеток. Типы нервных клеток, подверженные такой опасности, имеют длинные и тонкие аксоны, лишенные миелина или мало миелинизированные. В то же время, нервные клетки с длинными и короткими аксонами, обособленными миелиновым футляром толстого калибра, оказываются защищенными от образования конгломератов α-синуклеина и сохраняют свою морфологическую и функциональную целостность в течение всего периода развития БП. Это означает, что нейрональные повреждения в мозге, возникающие в процессе БП, совсем не случайны, а подчиняются определенным закономерностям.
Рассматривая внутриклеточные включения белка α-синуклеина в качестве основного индикатора стадий БП (в сочетании с анализом ряда патологических, анатомических и клинических данных), сотрудники Института клинической нейроанатомии (Фракфурт-на-Майне, Германия) разработали новую классификацию стадий нейродегенеративного процесса, описывающую не только клинические, но и доклинические, или премоторные стадии БП. Согласно гипотезе, тельца Леви с включенными в них агрегатами неправильно свернутого белка α-синуклеина развиваются последовательно, начинаясь с предрасположенных к ним определенных групп нейронов, и продвигаются в топографически предсказуемой последовательности в другие области серого вещества, уязвимые для дегенерации. Показатели процесса нейродегенерации в динамике 6-и стадий БП (рис. 3) ранее изложены в обзоре. Кратко резюмировать данные франкфуртской группы, положенные в основу новой классификации, можно следующим образом.
1. БП относят к заболеваниям, именуемым как «синуклеинопатии», в основе которых лежит образование внутриклеточных включений белка α-синуклеина в виде телец и нейритов Леви.
2. В патологию Леви при БП вовлечены предрасположенные к поражению типы нервных клеток, которые имеют длинные и тонкие аксоны, лишенные миелина или мало миелинизированные.
3. На доклинических, премоторных стадиях (стадии 1 и 2), когда классические для БП моторные симптомы отсутствуют, патология Леви ограничивается серым веществом в двух областях — продолговатом мозге и покрышке варолиева моста, а также в обонятельной луковице и переднем обонятельном ядре.
4. На поздней доклинической и ранней клинической стадиях (стадии 3 и 4), когда появляются первые моторные симптомы, фокусом сначала незначительной, а затем все более существенной патологии Леви становятся черная субстанция и ядра серого вещества в среднем и переднем мозге.
5. На поздних клинических стадиях (стадии 5 и 6) процесс захватывает области новой коры и болезнь проявляется в полном клиническом объеме; типичной чертой этих стадий являются когнитивные нарушения и деменция.

Необходимо отметить, что ряд положений новой классификации остаются дискуссионными и требуют уточнений. Образование телец и нейритов Леви обнаружено при различных заболеваниях, рассматриваемых как синуклеинопатии (см. табл. 1), а также при прогрессирующем надъядерном параличе и болезнях Пика и Альцгеймера. Например, при иммуногистохимическом исследовании мозга 260 пожилых людей выявлено наличие телец Леви не только при БП и деменции с тельцами Леви, но и примерно в 50% случаев диагноза болезни Альцгеймера и у 30% пожилых людей без нейропсихиатрических нарушений (контроль); данные сочетались с клиническим обследованием и были подтверждены аутопсией. Патология Леви встречается при болезни Альцгеймера, не сопровождающейся симптомами паркинсонизма.
Трудно дать убедительное объяснение приведенных выше удивительных данных об одновременном начале образования агрегаций Леви в двух, отдаленных друг от друга, областях — в продолговатом мозге/покрышке варолиева моста и обонятельной луковице/переднем обонятельном ядре. Авторы новой классификации выстраивают между этими областями красивую нейронную сеть из маломиелинизированных аксонов, но «строительного материала» недостаточно. Чрезвычайно ответственным является положение гипотезы об отсутствии патологии Леви в черной субстанции на доклинических стадиях 1 и 2.
Ряд авторов обнаружили отклонения от прогнозируемого гипотезой франкфуртской группы каудально-рострального направления α-синуклеиновой патологии и полагают, что они могут быть связаны с генетической предрасположенностью или сопутствующими заболеваниями. Эти данные позволяют предполагать, что путь, описанный в работах, не является единственным путем распространения в мозге α-синуклеиновой патологии. Объяснением этих несоответствий может стать предположение о распространении патологии не только в ретроградном, но и антероградном направлении. Кроме того в работе было отмечено, что пациенты с началом БП в более раннем возрасте и с длительным течением имели патологию, соответствующую франкфуртской модели, а пациенты с более поздним наступлением БП и коротким течением болезни — не соответствующую ей.
Следовательно, основные сомнения и уточнения касаются каудально-рострального направления нейродегенеративного процесса и той последовательности включения в него различных областей мозга, которую предопределяет гипотеза о стадиях БП. Роль накопления агрегаций α-синуклеина в патогенезе БП признается большинством исследователей.
Читайте также
Услуги гидроизоляции — внешняя или внутренняя?

Трудовые споры: как добиться справедливости от недобросовестного работодателя

Трудовые отношения — это тонкая материя, полная нюансов и правовых
Как отличить брендовые очки от подделки

Брендовые солнцезащитные очки — это не только модный аксессуар, но
Дизайн встроенной кухни: как оптимизировать пространство

Несмотря на большое разнообразие готовой (типовой) мебели, мебель на заказ
Михаил Владимирович Мишустин: отличный управленец и экономист

Михаил Владимирович Мишустин — выдающийся российский государственный и политический деятель,
Самые популярные рецепты пиццы: идеальное сочетание ингредиентов для настоящего гурмана

Пицца – это одно из наиболее популярных блюд в мире,
-

Лето – это время, когда дети, закончив учебный год, уходят
Как получить гражданство Бельгии и что оно дает?

Бельгия, расположенная в сердце Европейского союза, по праву считается одним
Осетинские пироги: вкусное и популярное блюдо с Кавказа

Когда начинать готовиться к ЕГЭ и ОГЭ 2024: полезные рекомендации

Начало нового учебного года часто становится временем повышенной тревожности как
На чем можно долететь до Мальдив? Регулярный рейс или аренда частного самолета?

Путешествие на Мальдивы — это мечта многих туристов. Острова, утопающие
Зубной имплантат: преимущества выбора при протезировании

Зубной имплантат – это современная технология, предоставляющая возможность восстановить утраченный
Яйцо шоколадное Kinder сюрприз: волшебство, которое завоевало сердца детей и взрослых

Яйцо Kinder сюрприз, безусловно, является одним из наиболее популярных шоколадных
Суши и пицца: почему они так популярны в службе доставки

Службы доставки еды становятся всё популярнее среди людей, желающих насладиться
Пептидные препараты: сущность и области применения

Пептидные препараты стали одним из важнейших направлений в современной медицине