Генетические факторы в патогенезе спорадической болезни Паркинсона
Изучение моногенных форм БП позволил выделить основные возможные патогенетические механизмы, которые могут быть также связаны с развитием спорадической формы БП. В первую очередь к ним относятся нарушение убиквитин-зависимой протеасомной деградации белков и дифференцировки дофаминергических нейронов, митохондриальная дисфункция и окислительный стресс, нарушение нормального функционирования синапса.
В большинстве случаев у больных развитие БП носит спорадический характер и определяется сложным взаимодействием между генетической конституцией организма и факторами внешней среды. В связи с этим возникает вопрос о том, какой вклад вносят в патогенез спорадической формы БП гены семейных форм заболевания и других генетических факторов.
Анализ генов моногенных форм БП, белки которых принимают участие в протеасомной деградации белков, показал, что все они могут быть вовлечены в патогенез спорадической формы БП. Так, в гене α-синуклеина выявлено несколько ассоциированных со спорадической формой БП интронных ОНП и динуклеотидный полиморфизм в промоторной области. Предполагают, что эти полиморфизмы могут влиять на транскрипционную активность гена и стабильность мРНК. Для другого гена, гена паркина, установлено, что гомозиготность, или компаундная гетерозиготность по мутациям в этом гене приводит к развитию заболевания у 5-10% больных со спорадической БП с ранним (до 50 лет) началом развития заболевания. Как уже говорилось выше, даже в гетерозиготном состоянии мутация в гене PARK2 может приводить к развитию заболевания или повышать чувствительность организма к действию факторов внешней среды. На последней стадии процессов деградации белков важную роль играют убиквитин-гидролазы, которые высвобождают свободный убиквитин из комплекса с белком. Одним из таких ферментов является убиквитин-карбоксигидролаза UCHL1, мутации в гене которого ведут к развитию семейной формы БП. Возможно, что полиморфизм этого гена может влиять на риск развития спорадической формы БП. Так, в настоящее время выявлена ассоциация между риском развития БП и частым полиморфизмом S18Y, расположенным в кодирующей области гена UCHL1, и установлено, что вариант 18Y снижает риск развития заболевания. В патогенезе БП могут также играть роль белки-мишени паркина, к которым относится синфилин-1. Как и α-синуклеин, он входит в состав телец Леви. В гене синфилина-1 выявлена мутация R621C у спорадических больных БП с поздним началом развития. Однако нельзя исключить, что данная мутация является редким нормальным полиморфизмом и не связана с развитием БП. Кроме перечисленных выше белков, в процессах убиквитин-зависимого протеолиза могут принимать участие и другие ферменты, которые также могут быть вовлечены в патогенез БП. Так, кроме убиквитина в протеасомной деградации принимают участие убиквитин-подобные белки NEDD8 и SUMO-1. Первый из них выявляется в тельцах Леви, тогда как белок SUMO-1 играет заметную роль в модификации белка DJ-1. Эти данные позволяют рассматривать гены этих белков как возможные кандидатные гены БП.
Роль нарушения процессов дифференцировки дофаминергических нейронов в патогенезе БП подтверждается выявлением мутаций в гене транскрипционного фактора Nurrl при одной из семейных форм БП. Ген Nurr1 кодирует транскрипционный фактор, необходимый для дифференцировки постмитотических дофаминергических нейронов и контролирующий экспрессию генов, белковые продукты которых связаны с синтезом и хранением дофамина, а именно — тирозингидроксилазы (ТН), транспортера дофамина (DAT), везикулярного транспортера моноаминов (VMAT2), декарбоксилазы ароматических аминокислот (AADC). Свой вклад в патогенез спорадической формы БП могут также вносить и другие транскрипционные факторы, необходимые для дифференцировки дофаминергических нейронов, такие как транскрипционные факторы Pitx3 и Lmx1b. В настоящее время в гене одного из них — фактора Pitex3, который специфичен для дофаминергических нейронов вентральной и вентромедиальной части черной субстанции, выявлены два полиморфизма, расположенные в первом нитроне и в непосредственной близости от 5′-конца гена Pitx3, ассоциированные со спорадической формой БП. Кроме того, в дифференцировке и поддержании нормального функционирования дофаминергических нейронов особую роль играют нейротрофический фактор мозгового происхождения (BDNF), нейротрофический фактор глиального происхождения (GDNF) и трансформирующий фактор роста α — TGF-α. Известно, что BDNF и GDNF влияют на обмен дофамина в черной субстанции. Однако только для одного из этих белков — BDNF, который регулирует экспрессию D3 рецептора дофамина на нейронах стриатума, был обнаружен полиморфизм V66M в гене BDNF, который ассоциирован со спорадической БП в японской популяции. Анализ же этого полиморфного варианта и других полиморфизмов в гене BDNF в европейских популяциях не выявил каких-либо ассоциаций с БП.
Снижение синтеза АТФ митохондриями может приводить к развитию БП по двум механизмам — через снижение уровня АТФ-зависимой протеасомной деградации белков и через повышение уровня свободных радикалов в клетке в условиях дисфункции электрон-транспортной цепи митохондрий. В свою очередь, повышение уровня свободных радикалов приводит к снижению активности протеасомного комплекса. Важная роль в патогенезе БП доказывается ранее описанными мутациями в гене белка DJ-1 при семейных формах заболевания. Белок DJ-1 играет роль протектора оксидантного стресса и тем самым защищает клетки от их повреждения свободными радикалами.
О возможной роли митоходриальной дисфункции в патогенезе БП говорит снижение активности комплекса I дыхательной цепи митохондрий в черной субстанции и стриатуме больных, а также выявление нескольких семей с БП, наследуемой по материнской линии. Об этом же также свидетельствует описанная в работе ассоциация между митохондриальными митотипами и риском развития спорадической БП. В ней показано, что у европейцев с митотипами J риск развития заболевания значительно ниже, чем у носителей самого частого в Европе митотипа Н. Другим ассоциированным с развитием БП вариантом митохондриальной ДНК является полиморфизм 10398G, влияющий на активность NADH-дегидрогеназы 3. Еще одним подтверждением роли митохондриального генома в БП являются эксперименты по получению цитоплазматических гибридов из клеток нейробластомы и митохондрий из черной субстанции больных паркинсонизмом. В таких гибридах наблюдалось образование включений, напоминающих по своей морфологии и составу тельца Леви. Повышение уровня свободных радикалов может быть обусловлено нарушением обмена железа. В черной субстанции у больных БП содержание железа повышено, причем наблюдается сдвиг соотношения между ионами двухвалентного и трехвалентного железа в сторону более окисленной формы. Свободные ионы железа приводят к образованию супероксид-аниона и перекисных соединений и индуцируют аутоокисление дофамина с образованием изохолина. Показано также, что повышение уровня железа стимулирует процесс образования агрегатов α-синуклеина.
Поддержание гомеостаза железа связано с рядом функциональных систем, таких как система транспорта железа в плазме крови (трансферрин и лактоферрин), система транспорта и хранения железа в клетке (ферритин, рецептор трансферина, HFE-белок), система обмена (фратаксин и церулоплазмин) и обратного транспорта (церулоплазмин и гемоксигеназа 1) железа в клетке. Гены, кодирующие белки перечисленных систем, могут рассматриваться как кандидатные гены БП. В последние годы была выявлена ассоциация некоторых из них с развитием заболевания.
Однако весь спектр возможных путей этиопатогенеза данной формы БП вышеперечисленными механизмами не исчерпывается. Наблюдаемая при БП симптоматика возникает в результате снижения синтеза дофамина в черной субстанции и в стриатуме и уменьшения его концентрации в стриатуме. Одной из причин этого может быть нарушение функционирования системы обмена дофамина — его синтеза, транспорта, синаптического выброса и рецепции.
При синтезе дофамина скорость процесса лимитируется реакцией превращения тирозина в ДОФА, катализируемой тирозингидроксилазой (ТН). В связи с этим ген ТН можно рассматривать как один из наиболее вероятных кандидатных генов БП. Однако до сих пор не обнаружено каких либо ассоциаций между полиморфными вариантами гена ТН и развитием заболевания. С другой стороны, большую роль в патогенезе БП играют белки, регулирующие активность (а-синуклеин) и транскрипцию гена (Nurrl) тирозингидроксилазы. В связи с этим можно предположить, что особое место в развитии БП может иметь нарушение синтеза кофактора тирозингидроксилазы — тетрагидробиоптерина (ВН4). В некоторых случаях мутации в гене ГТФ циклогидролазы (GCH1), первого фермента цикла биосинтеза ВН4, приводит к развитию паркинсон-подобного фенотипа, неклассической картине ДОФА-зависимой торзионной дистонии. Ген фермента SRR, катализирующий последний этап биосинтеза ВН14, картирован в области локуса PARK3, и не исключено, что именно мутации в этом гене ответственны за PARK3-форму БП.
Уровень дофамина в клетке зависит также от скорости его деградации и под действием моноаминоксидазы В (МАО-В), и активности дофамин-β-гидролазы, использующей дофамин при синтезе норадреналина. Для всех генов этих ферментов выявлены ассоциации различных полиморфных вариантов с развитием БП.
Не исключена также роль в развитии БП системы обратного транспорта дофамина и рецепторов дофамина. Наиболее заметно при БП изменяется экспрессия рецепторов дофамина D3, уровень которого снижается на 40-45% в области nucleus accumbens и покрышке, и D4, уровень которого в покрышке возрастает на 15%. Однако в настоящее время нет убедительных данных о наличии ассоциации между генами рецепторов дофамина и БП.
Развитие БП может быть связано с токсическим действием различных ксенобиотиков. В связи с этим важную роль в патогенезе заболевания могут играть ферменты системы детоксикации. Проводится активное изучение как ферментов первой стадии этого процесса, при которой происходит активация ксенобиотиков, в первую очередь белков суперсемейства цитохрома Р450 (CYP2E1, CYP2D6), так и ферментов второй стадии, заключающейся в детоксикации эндогенных и экзогенных токсинов, — глутатион-S-трансфераз (GSTT1, GSTM1, GSTP1) и N-ацетилтрансферазы (NAT2). Однако до настоящего времени не выяснено, могут ли ферменты первой стадии детоксикации играть какую-либо роль в развитии БП. Более интересные данные получены при изучении ферментов второй стадии. Так, для аллельных вариантов гена GSTP1 (I104V, A113V) установлено наличие ассоциации с развитием БП, в то время как для «нулевых» аллельных вариантов генов GSTT1, GSTM1 не выявлено какой-либо связи с развитием заболевания. Результаты метаанализа данных, полученных в различных этнических группах, для фенотипа медленных метаболизаторов фермента NAT2, к которому приводят различные мутации в гене NAT2, показали, что данный фенотип ассоциирован с развитием БП.
Кроме того, к настоящему времени проведено более трех десятков полногеномных исследований ассоциаций для БП. Необходимо отметить, что ассоциации, полученные в первых исследованиях (работы Д.Н. Марагано и др., Т.Г. Лесник и др., Х.С. Фанг и др.) не были воспроизведены в других более поздних работах. Это связано с тем, что данные, полученные в первых работах, не являются достоверными? согласно позднее появившемуся критерию достоверности для полногеномных ассоциативных исследований. Так как при полногеномном анализе ассоциаций проводится одновременный анализ большого числа ОНП, обычный уровень достоверности (р<0,05) приводит к появлению большого числа ложноположительных ассоциаций. Было вычислено новое значение достоверности, которое учитывает одновременный анализ 1 миллиона ОНП, путем введения поправки на многократное тестирование (аналог поправки Бонферрони). На данный момент считается, что отдельный ОНП может считаться ассоциированным с развитием заболевания при p<5*10в-8.
Большое число работ по полногенномному анализу ассоциаций позволило провести также метаанализ данных, полученных в различных полногеномных исследованиях ассоциаций при БП (табл. 2).
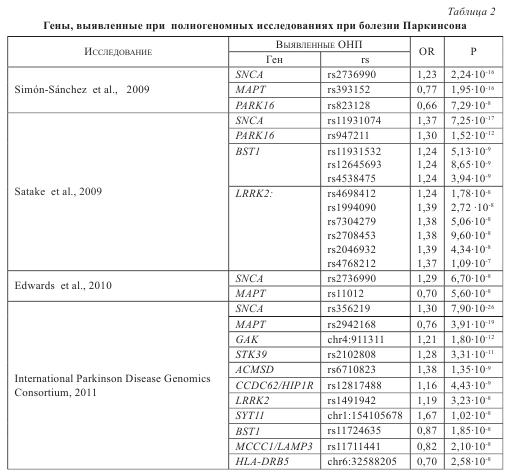
Суммируя данные по проведенным на сегодняшний день полногенным ассоциативным исследованиям, можно сделать вывод, что только для двух генов -SNCA и МАРТ — были выявлены строгие ассоциации с БП практически во всех исследованиях. Для генов BST1, LRRK2, GAK-DGKQ, HLA-DRA и локуса PARK16 были выявлены ассоциации лишь в ряде работ и не были подтверждены большинством исследователей.
Основной причиной таких различий в получаемых результатах при полногенных ассоциативных исследованиях является необходимость использования очень больших по размеру выборок больных. В результате в исследованиях используются гетерогенные выборки — как клинически, так и этнически. Кроме того, ассоциации отдельных ОНП с БП могут быть специфичны для разных популяций.
В России в настоящее время начаты работы по верификации данных, полученных при полногеномном анализе ассоциаций в европейских популяциях. Проведен анализ однонуклеотидного полиморфизма в гене WNT3 (rs415430), расположенном рядом с геном МАРТ, и отобранного по результатам полногеномных исследований ассоциаций. Однако ассоциация данного полиморфизма с риском развития БП в российской популяции не была выявлена.
Читайте также
Особенности сантехники из Италии: почему стоит переплатить

Отдых в Джемете — солнце, море, пляж

Джемете — это живописный район на побережье Черного моря, который
Школа профессионального макияжа: искусство создания идеального образа

-

Дорамы – популярный жанр телевизионного и кинематографического искусства, который нашел
Правильный выбор ткани — половина успеха вашего образа

Разработка брендбука: почему это важно

Услуги гидроизоляции — внешняя или внутренняя?

Трудовые споры: как добиться справедливости от недобросовестного работодателя

Трудовые отношения — это тонкая материя, полная нюансов и правовых
Как отличить брендовые очки от подделки

Брендовые солнцезащитные очки — это не только модный аксессуар, но
Дизайн встроенной кухни: как оптимизировать пространство

Несмотря на большое разнообразие готовой (типовой) мебели, мебель на заказ
Михаил Владимирович Мишустин: отличный управленец и экономист

Михаил Владимирович Мишустин — выдающийся российский государственный и политический деятель,
Самые популярные рецепты пиццы: идеальное сочетание ингредиентов для настоящего гурмана

Пицца – это одно из наиболее популярных блюд в мире,
-

Лето – это время, когда дети, закончив учебный год, уходят
Как получить гражданство Бельгии и что оно дает?

Бельгия, расположенная в сердце Европейского союза, по праву считается одним
Осетинские пироги: вкусное и популярное блюдо с Кавказа
