Состояние нигростриатной дофаминергической системы на моделях доклинической и ранней клинической стадий болезни Паркинсона
Как упоминалась раннее, длительное бессимптомное течение БП связано отчасти с развитием компенсаторных процессов в мозге, истощение которых можно рассматривать как триггер перехода из доклинической в клиническую стадию БП. Для поиска таких триггеров были использованы модели доклинической и ранней клинической стадии БП с дальнейшим сравнением полученных результатов.
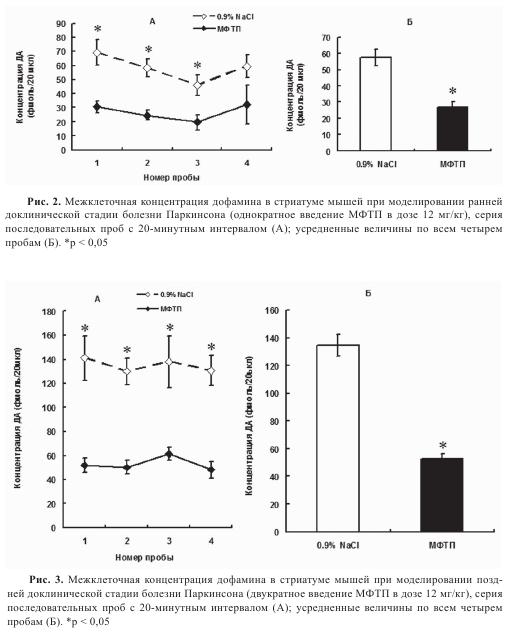
Межклеточная концентрация ДА в стриатуме. Ключевым показателем функциональной активности нигростриатной ДА-ергической системы является межклеточная концентрация ДА в стриатуме, т.е. в той области мозга, где он обеспечивает межклеточную сигнализацию в цепочке нейронов, включенной в регуляцию моторного поведения. Действительно, межклеточное содержание ДА — это интегративный показатель, который отражает синтез нейротрансмиттера, его выделение из нейрона и обратный захват, деградацию в межклеточном пространстве. Определение межклеточной концентрации ДА и его метаболитов в стриатуме осуществляли на моделях ранней и поздней доклинической стадиях и на ранней клинической стадии БП с помощью микродиализа. На ранней досимптомной стадии у мышей (1×12 мг/кг МФТП) обнаружено уменьшение вдвое (на 54,3%) межклеточной концентрации ДА по сравнению с контролем (рис. 2). Интересно, что на этой стадии соизмеримы изменения содержания ДА в аксонах и в межклеточном пространстве в стриатуме. В отличие от ДА, концентрации L-ДОФА — предшественника синтеза ДА, а также ДОФУК и гомованилиновой кислоты (ГВК) — продуктов деградации ДА, не изменялись по сравнению с контролем.
На поздней досимптомной стадии (2×12 мг/кг МФТП) содержание ДА в межклеточном пространстве уменьшилось еще на 7% (в целом на 63%) по сравнению с ранней симптомной стадией (рис. 3).
На модели ранней симптомной стадии паркинсонизма (4×12 мг/кг МФТП) наблюдается уменьшение межклеточной концентрации ДА в стриатуме еще в большей степени, чем на модели досимптомной стадии — на 75% (рис. 4).
Из сопоставления полученных данных следует, что в процессе развития паркинсонизма от ранней досимптомной стадии до ранней симптомной стадии наблюдается нарастающее снижение межклеточного уровня ДА в стриатуме (рис. 5).
Полученные данные по межклеточному содержанию ДА полностью коррелируют с внутриклеточным содержанием нейротрансмиттера в аксонах стриатума (данные ВЭЖХ с ЭД) на исследуемых стадиях паркинсонизма у мышей, что подтверждает тот факт, что симптомы паркинсонизма появляются после снижения действующей (межклеточной) концентрации ДА в стриатуме более чем на 70%.
Для того чтобы определить, какие именно компенсаторные процессы поддерживают ДА выше порогового значения на досимптомной стадии и срыв каких механизмов приводит к снижению уровня этого нейротрансмиттера на симптомной, были количественно охарактеризованы следующие показатели: синтез, деградация, выделение и обратный захват ДА, а также уровень мРНК Д2 рецепторов как показатель чувствительности клеток-мишеней к ДА. Учитывая то, что для каждой стадии паркинсонизма было определено количество тел и терминалей аксонов ДА-ергических нейронов, некоторые параметры, например содержание ДА, можно было пересчитать на отдельный нейрон — тело нейрона в ЧС и терминаль аксона в стриатуме.
Синтез и деградация дофамина. Уровень синтеза ДА оценивали по содержанию мРНК и белка ТГ, а также по активности фермента в телах и терминалях аксонов ДА-ергических нейронов соответственно в ЧС и стриатуме. На досимптомной стадии паркинсонизма у мышей в ЧС, несмотря на потерю почти 30% тел ДА-ергических нейронов, уровень мРНК ТГ остается прежним (см. табл. 4), что свидетельствует об увеличении экспрессии гена ТГ в отдельном нейроне. Интересным представляется тот факт, что содержание ТГ (белка) в отдельном нейроне при этом не меняется (ТГ снижено во всей области ЧС на 45%). Такое расхождение в уровне мРНК и белка ТГ свидетельствует о разбалансировке процессов транскрипции и трансляции. В телах ДА-ергических нейронов на досимптомной стадии происходит увеличение транскрипции и либо снижение трансляции, либо увеличение скорости деградации фермента. Эта закономерность была отмечена ранее при моделировании БП на крысах с помощью 6-ГДА.

На ранней симптомной стадии наблюдалась противоположная картина — при потере 43% тел ДА-ергических нейронов в ЧС содержание мРНК ТГ также было снижено на 50%, что свидетельствует о сохранении экспрессии гена ТГ в отдельных нейронах на уровне контроля. При этом содержание ТГ в ЧС не менялось, а при пересчете на отдельный ДА-ергический нейрон даже увеличилось на 29%, что свидетельствует о компенсаторном увеличении скорости трансляции ТГ в выживших нейронах на ранней симптомной стадии паркинсонизма у мышей при неизменном уровне транскрипции мРНК ТГ.
На поздней досимптомной и ранней симптомной стадиях паркинсонизма содержание ТГ в терминалях аксонов ДА-ергических нейронов в стриатуме снизилось в равной степени — соответственно на 28% и 26%. Сниженное содержание ТГ в отдельных терминалях аксонов при неизменном (на досимптомной стадии) или, более того, повышенном (на ранней симптомной стадии) уровне фермента в телах нейронов может свидетельствовать либо об увеличенной деградации ТГ в аксонах, либо о нарушении антероградного аксонального транспорта, что более вероятно. Действительно, было показано, что МФП+ увеличивает скорость денеин-зависимого ретроградного транспорта и снижает скорость быстрого кинезин-зависимого антероградного транспорта in vitro на аксоне гигантского кальмара через активацию каспаз и протеинкиназы С.
Наиболее важным показателем секреторной активности ДА-ергических нейронов является активность ТГ, которая и определяет скорость синтеза ДА. В ЧС ни на досимптомной, ни на ранней симптомной стадиях изменений активности ТГ не было обнаружено, тогда, как в отдельных выживших нейронах активность ТГ повышена. Полученные данные хорошо согласуются с данными по уровню ДА в ЧС, который не менялся ни на одной из моделей паркинсонизма у мышей, что в совокупности свидетельствует о неизменном уровне синтеза ДА.
В стриатуме на моделях досимптомной и ранней симптомной стадий паркинсонизма у мышей произошло одинаковое снижение активности ТГ — на 47% и 48%, в то время как в отдельной терминали ДА-ергичского аксона в стриатуме происходило увеличение активности этого фермента соответственно на 28% и 59% по отношению к контролю (см. табл. 4). Учитывая то, что активность ТГ отражает скорость синтеза ДА, увеличение этого параметра на фоне неизменного уровня ДА в отдельной терминали аксона на досимптомной стадии свидетельствует о возможном увеличении выделения ДА или снижении его обратного захвата. Также полученные данные могут отражать снижение активности второго фермента синтеза ДА — декарбоксилазы ароматических L-аминокислот (ДАА), что было показано при БП. По-видимому, эти процессы усугубляются на ранней симптомной стадии паркинсонизма по сравнению с досимптомной стадией. Из приведенных данных следует, что триггером нарушения двигательной функции у мышей на симптомной стадии является не снижение скорости синтеза ДА, а увеличение его обратного захвата или снижение скорости выделения.
Учитывая то, что содержание ДА в нейронах и в межклеточном пространстве отчасти определяется активностью ферментов деградации ДА, на моделях досимптомной и ранней симптомной стадий паркинсонизма оценивали активность моноаминоксидазы (МАО) по содержанию продуктов деградации — ДОФУК и ГВК. МАО — внутриклеточный фермент, представленный у млекопитающих двумя изоформами — МАО А и МАО Б. МАО А содержится только в нейронах, в то время как МАО Б в основном в астроцитах. У грызунов ДА метаболизируется в основном МАО А, в то время как МАО Б вносит незначительный вклад в этот процесс. МАО А метаболизирует ДА до ДОФУК, после чего подключается второй фермент деградации ДА — катехол-О-метилтрансфераза (КОМТ), осуществляющий метилирование ДОФУК до ГВК. Также деградация ДА может происходить в обратном порядке, когда КОМТ вначале метаболизирует ДА до 3-метокситирамина, который деградирует до ГВК при участии МАО. Также был оценен интегральный показатель синтеза и деградации ДА — оборота ДА «turnover» (ДОФУК/ДА и ГВК/ДА).
Активность МАО А в ЧС не изменилась ни на досимптомной стадии паркинсонизма у мышей, ни на ранней симптомной стадии. Более того, не было обнаружено изменений в деградации ДА, определяемой по уровню ДОФУК и ГФК, а также по показателю «turnover», в ЧС ни на одной из изученных стадий (см. табл. 4). При этом полученные данные хорошо согласуются с данными о неизменном уровне ДА и активности ТГ в ЧС на досимптомной и ранней симптомной стадиях паркинсонизма.
В стриатуме активность МАО А на досимптомной стадии также не изменилась по сравнению с контролем, в то время как при переходе в симптомную стадию увеличилась в 2,5 раза. При пересчете на отдельные терминали ДА-ергических аксонов в стриатуме на симптомной стадии активность МАО А увеличилась на 678% относительно контроля. ГВК и ДОФУК были снижены примерно в равной степени на обеих стадиях паркинсонизма, в то время как показатель «turnover» на досимптомной стадии был увеличен на 50%, а на ранней симптомной до 250%, что хорошо согласуется с увеличенной активности МАО А. Таким образом, наше предположение о снижении ДА в стриатуме на ранней симптомной стадии до порогового уровня при сохранении скорости синтеза нейротрансмиттера за счет увеличения скорости его деградации подтвердилось.
Исследование активности МАО Б представляет особый интерес, т.к. именно этот фермент метаболизирует МФТИ до МФП+. Изменение активности МАО Б было обнаружено только на досимптомной стадии паркинсонизма у мышей в стриатуме. Известно, что МФТИ вызывает необратимое ингибирование МАО Б, как это было показано в опытах in vitro на митохондриях крыс. В случае изучения компенсаторных механизмов и поиска триггеров перехода из досимптомной стадии БП в симптомную изменения всех параметров изучают на отдаленных сроках после введения МФТИ, в нашей работе — через 14 дней после введения МФТИ. За это время должен был пройти полный цикл обновления функциональных белков, включая МАО Б, поэтому ингибирование активности МАО Б с помощью МФТИ представляется маловероятным. Такое снижение активности МАО Б можно рассматривать как компенсаторный механизм, который направлен на увеличение межклеточного содержания ДА. С другой стороны, МАО Б принимает участие в деградации не только ДА, но и других моноаминов. Изменение его активности может быть направлено на изменение метаболизма других нейротрансмиттеров, например, широко представленного в стриатуме серотонина.
Таким образом, к компенсаторным процессам можно отнести увеличенный синтез ДА, который возрастает при переходе в симптомную стадию. Триггером перехода является увеличение активности МАО А, т.е. деградации нейротрансмиттера, что приводит к снижению уровня ДА ниже порогового значения на симптомной стадии.
Выделение и обратный захват дофамина. К гипотетическим компенсаторным механизмам, которые поддерживают ДА на необходимом уровне в стриатуме, можно отнести увеличение выделения и снижение обратного захвата нейротрансмиттера, что приводит к более длительному «пребыванию» ДА в синаптической щели. С другой стороны, срыв одного из этих механизмов может приводить к снижению межклеточного содержания ДА, т.е. являться триггером перехода из досимптомной стадии в раннюю симптомную стадию паркинсонизма.
Скорость спонтанного и К+-стимулированного выделения ДА в ответ на калиевую деполяризацию мембраны, определяли in vitro путем перфузии срезов ЧС и стриатума с дальнейшей оценкой содержания ДА в перфузированной среде (см. табл. 4). Ни спонтанное, ни стимулированное выделение ДА в ЧС не изменилось ни на одной из моделей БП. В отдельных выживших ДА-ергических нейронах на досимптомной стадии спонтанное выделение также не изменялось, в то время как стимулированное выделение повысилось на 37%. На ранней симптомной стадии увеличились оба показателя — в телах нейронов на 77% спонтанное выделение ДА и на 63% стимулированное выделение. Таким образом, при переходе из досимптомной в симптомную стадию компенсаторно возрастает спонтанное выделение ДА из тел нейронов в ЧС.
Спонтанное выделение ДА в стриатуме на досимптомной и ранней симптомной стадии паркинсонизма снижено соответственно на 40% и 56%. При оценке спонтанного выделения ДА на терминаль аксона в стриатуме не обнаружено различий между опытом и контролем, что свидетельствует об отсутствии компенсаторного увеличения этого показателя. Иная картина наблюдается при К+-стимулированном выделении ДА в стриатуме. На досимптомной стадии паркинсонизма происходит увеличение стимулированного выделения ДА на 29%, в то время как на ранней симптомной происходит значительное снижение — на 28% относительно контроля. При этом стимулированное выделение ДА из отдельных терминалей аксонов на досимптомной стадии увеличено на 216% по сравнению с контролем, а на симптомной стадии составляет 125%. Уменьшение этого показателя при переходе из одной стадии в другую можно рассматривать как триггер нарушения моторного поведения у животных.
Скорость обратного захвата ДА оценивали ex vivo путем статической инкубации срезов ЧС и стриатуме с Н3-ДА. Скорость обратного захвата ДА в ЧС не меняется ни на одной из рассматриваемых моделей, что хорошо согласуется с неизменным уровнем выделения нейротрансмиттера. В стриатуме наблюдается небольшое, но статически достоверное увеличение захвата Н3-ДА на досимптомной стадии, в то время как на ранней симптомной стадии изменений по сравнению с контролем нет. Следовательно, при переходе из досимптомной в симптомную стадию происходит минимальное снижение обратного захвата ДА в стриатуме, что является компенсаторным механизмом поддержания ДА в синаптической щели.
Из сопоставления данных по стимулированному выделению ДА из терминалей ДА-ергических аксонов в стриатуме на досимптомной и ранней симптомной стадиях паркинсонизма следует, что выделение ДА снижается, что может быть одной из причин перехода из одной стадии в другую. На досимптомной стадии паркинсонизма показано увеличение обратного захвата ДА в стриатуме на 5% от уровня в контроле (статистически достоверно). В то же время на симптомной стадии обратный захват снижается до контрольного уровня, что также можно рассматривать как триггер нарушения моторного поведения. Полученные данные в целом свидетельствуют о разбалансированности процессов, которые регулируют межклеточное содержание ДА в стриатуме.
Чувствительность нейронов-мишеней к дофамину в стриатуме. Было показано, что как у людей, так и при моделировании БП на мышах снижение содержание ДА в стриатуме приводит к увеличению экспрессии мРНК Д2-рецепторов и их плотности, но без изменений в аффинности к ДА. По нашим данным, уровень мРНК Д2-рецепторов с стриатуме не меняется по сравнению с контролем ни на досимптомной стадии паркинсонизма у мышей, ни на ранней симптомной, несмотря на значительное снижение ДА (75-процентное снижение на ранней симптомной стадии). Это может свидетельствовать о том, что такого снижения ДА недостаточно для активации этого компенсаторного процесса (см. табл. 4).
Таким образом, можно заключить, что к компенсаторным механизмам, которые предотвращают появления нарушений моторного поведения на досимптомной стадии, а также направлены на компенсацию низкого уровня ДА на ранней симптомной стадии, относится увеличение скорости синтеза ДА. К триггерам перехода из досимптомной стадии в раннюю симптомную стадию паркинсонизма можно отнести: снижение выделения и увеличение обратного захвата ДА на уровне аксонов с увеличением активности МАОА, что приводит к снижению содержания ДА в ткани стриатума и в межклеточном пространстве. Отсюда можно заключить, что для возможной коррекции нарушения двигательной функции можно использовать вещества, которые повысили бы содержание ДА в стриатуме выше порогового уровня (25%) за счет увеличения скорости выделения ДА в межклеточное пространство, снижения обратного захвата нейротрансмиттера и снижения скорости деградации ДА.
Читайте также
Услуги гидроизоляции — внешняя или внутренняя?

Трудовые споры: как добиться справедливости от недобросовестного работодателя

Трудовые отношения — это тонкая материя, полная нюансов и правовых
Как отличить брендовые очки от подделки

Брендовые солнцезащитные очки — это не только модный аксессуар, но
Дизайн встроенной кухни: как оптимизировать пространство

Несмотря на большое разнообразие готовой (типовой) мебели, мебель на заказ
Михаил Владимирович Мишустин: отличный управленец и экономист

Михаил Владимирович Мишустин — выдающийся российский государственный и политический деятель,
Самые популярные рецепты пиццы: идеальное сочетание ингредиентов для настоящего гурмана

Пицца – это одно из наиболее популярных блюд в мире,
-

Лето – это время, когда дети, закончив учебный год, уходят
Как получить гражданство Бельгии и что оно дает?

Бельгия, расположенная в сердце Европейского союза, по праву считается одним
Осетинские пироги: вкусное и популярное блюдо с Кавказа

Когда начинать готовиться к ЕГЭ и ОГЭ 2024: полезные рекомендации

Начало нового учебного года часто становится временем повышенной тревожности как
На чем можно долететь до Мальдив? Регулярный рейс или аренда частного самолета?

Путешествие на Мальдивы — это мечта многих туристов. Острова, утопающие
Зубной имплантат: преимущества выбора при протезировании

Зубной имплантат – это современная технология, предоставляющая возможность восстановить утраченный
Яйцо шоколадное Kinder сюрприз: волшебство, которое завоевало сердца детей и взрослых

Яйцо Kinder сюрприз, безусловно, является одним из наиболее популярных шоколадных
Суши и пицца: почему они так популярны в службе доставки

Службы доставки еды становятся всё популярнее среди людей, желающих насладиться
Пептидные препараты: сущность и области применения

Пептидные препараты стали одним из важнейших направлений в современной медицине