Роль предшественника амилоидного пептида и его метаболитов в патогенезе болезни Альцгеймера
В связи с увеличением средней продолжительности жизни старческая деменция, в частности, обусловленная развитием болезни Альцгеймера (БА), приобретает все более широкий характер и становится одной из главных причин тяжелой степени инвалидности людей пожилого возраста. В связи с этим в развитых странах исследования патогенеза БА являются одним из приоритетных направлений фундаментальных наук и медицины. Однако, несмотря на интенсивные исследования молекулярных основ патогенеза БА и существенный прогресс в понимании механизмов развития данного заболевания, все еще нет адекватных методов его профилактики и лечения. Это обусловлено тем, что сформулированная более 20 лет назад гипотеза амилоидного каскада, которая связывает патогенез БА с уровнем накопления в ткани мозга агрегатов β-амилоидного пептида (Аβ), базируется, в основном, на изучении семейных форм БA, вызванных мутациями определенных генов, хотя в основном они представлены редкими моногенетическими формами, поражающими отдельные семьи в различных районах мира. Для этих заболеваний характерными являются мутации генов белка предшественника амилоидного пептида (APP) или пресенилинов PSN1 и PSN2 (рис. 1). В то же время, БA имеет сложную этнологию, зависящую как от генетических, так и от разнообразных внешних факторов, и у пожилых людей чаще всего наблюдается спорадическая форма БA, не связанная с мутациями данных генов, но, тем не менее, обусловленная изменениями метаболизма амилоидного пептида под действием разных факторов. К их числу относятся гипоксия и ишемия мозга, окислительный стресс, травмы и воспалительные процессы в ткани мозга, снижающие его способность избавляться от токсических молекул по периваскулярному пути, а также экспрессия аллелей ряда генов, предрасполагающих к развитию БA (в частности, аполипопротеина АРОЕ) (см. рис. 1). Как было показано рядом авторов ApoE связывается с Ab и входит в состав как сенильных бляшек, так и нейрофибриллярных клубков и его экспрессия повышена при БA. Дальнейшие исследования подтвердили, что у носителей одной из трех аллелей АРОЕ, а именно APOE ε4, существенно увеличен риск развития BA. Так, у гетерозиготных пациентов он возрастает в 3 раза, а у гомозиготных — в 15 раз. Напротив, наличие аллели АРОЕ ε2 имеет защитный характер.
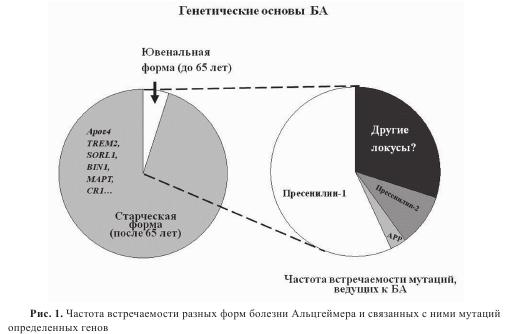
Исследования последних лет с использованием полногеномного поиска ассоциаций (Genome-Wide Association Study — GWAS), который требует выборок в сотни или тысячи индивидов и обязательной репликации ассоциаций на других популяциях, свидетельствуют о возможном вкладе большого числа генов в патогенез старческой формы БА, увеличивая предрасположенность к развитию этого заболевания. К ним помимо APOE относятся гены CLU, PICALM, CR1, BIN1, ABCA7, MS4A6A, CD33, ЕРНА1, SORL1 и многие другие. Все перечисленные выше факторы и мутации, способствующие накоплению в ткани мозга токсических форм амилоидного пептида, приводят к гибели нервных клеток и нарушению когнитивных функций. С другой стороны, исследование когорты пожилых людей в Исландии с отсутствием признаков деменции выявило наличие мутации в гене APP (А673Т), препятствующей развитию когнитивного дефицита и БА. Это открывает новое перспективное направление в исследовании эндогенных защитных механизмов, которые могут способствовать увеличению продолжительности активной деятельности мозга.
Амилоидный пептид (Аβ), представляющий собой полипептид с молекулярным весом около 4 кДа и состоящий из 36-43 аминокислотных остатков, образуется из молекулы белка предшественника амилоидного пептида, βAPP (β-Amyloid Precursor Protein), или просто АРР, в результате протеолиза под действием специфических протеиназ, а именно β- и γ-секретаз. β-секретаза принадлежит к классу аспартамовых протеаз, в то время как γ-секретаза представляет собой сложный многокомпонентный комплекс, в состав которого входят белки пресенилины PS1 и PS2, никастрин, APH1 и PEN2. Aβ обладает высоковыраженными фибрилогенными свойствами, и его олигомеры являются токсичными для нервных клеток, вызывая дегенерацию и гибель нейронов. Другим характерным морфологическим признаком БА является нарушение цитоскелета нервных клеток и накопление в них нейрофибриллярных клубков (НФК), состоящих в основном из нерастворимых филаментов гиперфосфорилированного белка тау. В нормальных физиологических условиях тау-протеин регулирует формирование и поддерживает структурную стабильность микротрубочек. В ходе развития БА наблюдается абнормальное гиперфосфорилирование тау, ведущее к дестабилизации и разрушению микротрубочек с последующим высвобождением тау-белка и его агрегации в спиралевидные филаменты. Несмотря на то что тау в основном локализуется в аксонах нервных клеток, его недавнее обнаружение в дендритных шипиках указывает на важную роль этого белка в процессах нейрональной пластичности, которая необходима для формирования памяти и реализации когнитивных функций. Накопление НФК при отсутствии Ар-патологии наблюдается при ряде других нейродегенеративных заболеваний, а частности, при фронтотемпоральной деменции, которая является второй наиболее часто встречающейся формой когнитивных растройств в возрасте до 65 лет. Как и в случае БА, развитие редких семейных форм этого заболевания обусловлено мутациями гена МАРТ, кодирующего тау-белок. При БА уровень накопления НФК коррелирует со степенью развития заболевания и когнитивным дефицитом, вызванным нарушением синаптической передачи и гибелью нейронов, в основном в коре и гиппокампе мозга. Хотя есть данные, свидетельствующие, что в ходе нейродегенеративного процесса сначала образуются амилоидные фибриллы, которые нарушают функции нервных клеток, а затем в них происходит формирование нейрофибриллярных клубков, в настоящее время все еще нет ясной концепции того, как эти два белка взаимодействуют на молекулярном уровне, приводя к патологии. Несмотря на то что при БA, в основном, имеет масто нарушение холинергической синаптической передачи, данные литературы свидетельствуют о том, что данная патология также сопровождается снижением содержания серотонина, норэпинефрина и других моноаминов.
До недавнего времени считалось, что накопление амилоидных фибрилл с последующим формированием склеротических бляшек в ткани мозга является необратимым процессом, однако сейчас уже не вызывает сомнений тот факт, что амилоидный пептид выводится из ткани мозга как по периваскулярным путям, так и подвергается протеолитическому расщеплению под действием широкого спектра ферментов. В связи с этим исследование механизмов освобождения мозга от избытка Ab сейчас считается одним из наиболее перспективных направлений для создания терапевтических средств, направленных на предотвращение и лечение БA. Согласно современным представлениям, метаболизм APP и Aβ является динамическим процессом, зависящим как от разнообразных внутренних факторов (генетических, клеточных, васкулярных), так и факторов окружающей среды (гипоксия, стресс, диета). Перечисленные факторы могут влиять как на формирование и накопление амилоидного пептида, так и на его деградацию и выведение из мозга. Поскольку частота проявлений БA в пожилом возрасте существенно повышается в связи с нарушением снабжения мозга кислородом при ишемии и инсультах, большое значение приобретают исследования роли гипоксии и ишемии мозга в процессах образования Аβ, а также в экспрессии как амилоид-образующих, так и амилоид-деградирующих ферментов.
Несмотря на всестороннее изучение процессов формировании токсичного амилоидного пептида, все еще нет ясного представления о биологической значимости его белка-предшественника — АРР, хотя накопленные к настоящему времени данные позволяют считать, что продукты его расщепления играют важную роль в нормальном функционировании нервной ткани. Так, например, Aβ в низких физиологических концентрациях является классическим нейропептидом и выполняет ряд важных функций, растворимые формы APP (sAPPα и sAPPβ) являются нейропротекторными, а образующийся под действием γ-секретазы C-концевой фрагмент APP (AICD) является транскрипционным фактором, регулирующим экспрессию ряда нейрональных генов, в том числе амилоид-деградирующей нейропептидазы неприлизин (НЕП). Дефицит активности НЕП и ряда других амилоид-деградирующих ферментов, способных расщеплять Aβ и препятствовать накоплению его токсических форм, вносит существенный вклад в развитие спорадической формы БА. Исследование механизмов регуляции НЕП при участии AICD показало, что этот процесс носит специфический для нервных клеток характер и зависит от уровня экспрессии нейрональной изоформы APP (APP695). В ходе активации экспрессии гена НЕП уровень связывания AICD с его промотором повышается и одновременно происходит снижение связывания с ним гистонедеацетилаз (в частности HDACI). Действие HDAC ингибиторов на клетки с низким уровнем экспрессии НЕП также приводит к увеличению связывания AICD с промотором гена НЕП и повышению его экспрессии. Поскольку уровень экспрессии и активности НЕП существенно снижается с возрастом, а также при гипоксии и ишемии мозга, приводя к развитию когнитивных расстройств и БА, направленное повышение уровня экспрессии НЕП в ткани мозга имеет большое практическое значение. В связи с этим поиск соединений, способных повышать уровень НЕП и снижать накопление амилоидного пептида, рассматривается как одно из перспективных направлений для создания средств профилактики БА. Развивая эти исследования, нами было показано, что введение животным антиконвульсивного препарата вальпроата натрия, который также является ингибитором гистондеацетилаз, приводит к повышению активности и экспрессии НЕП в коре и гиппокампе мозга и снижению когнитивных нарушений. Недавно нами было выявлено, что белок транстиретин (ТТР), который способен связывать и выводить Aβ из ткани мозга, регулируется в нейрональных клетках сходным с НЕП образом, а инкубация клеток нейробластомы с вальпроатом натрия приводит к одновременному повышению уровня экспрессии НЕП и TTP и снижению содержания Aβ. Полученные данные имеют большое практическое значение и позволяют надеяться на создание терапии, направленной на повышение содержания и активности амилоид-выводящих белков, уровень которых снижается при старении и развитии БА.
Как отмечалось на недавних международных совещаниях по патогенезу нейродегенеративных заболеваний и БА, разработка путей повышения способности мозга справляться с изменениями его гомеостаза и накоплением амилоидного пептида за счет активации ферментов его деградации, а также эффективности его удаления из нервной ткани, является на данный момент наиболее приоритетным и перспективным направлением исследований. С другой стороны, метаанализ данных литературы, а также развитие современных методов генотипирования и карт однонуклеотидных полиморфизмов (SNP) увеличивает список генов, повышающих риск развития БА или, напротив, предотвращающих это заболевание, что позволяет надеяться на создание более эффективных методов ранней диагностики и средств терапии этого заболевания. Накопленный опыт свидетельствует, что за более чем два десятилетия теория амилоидного каскада, лежащая в основе наших представлений о патогенезе БА, все еще не утратила своей значимости и, наоборот, позволяет критически относиться к полученному массиву данных и фокусировать внимание исследователей на наиболее перспективных направлениях исследований.
Читайте также
Услуги гидроизоляции — внешняя или внутренняя?

Трудовые споры: как добиться справедливости от недобросовестного работодателя

Трудовые отношения — это тонкая материя, полная нюансов и правовых
Как отличить брендовые очки от подделки

Брендовые солнцезащитные очки — это не только модный аксессуар, но
Дизайн встроенной кухни: как оптимизировать пространство

Несмотря на большое разнообразие готовой (типовой) мебели, мебель на заказ
Михаил Владимирович Мишустин: отличный управленец и экономист

Михаил Владимирович Мишустин — выдающийся российский государственный и политический деятель,
Самые популярные рецепты пиццы: идеальное сочетание ингредиентов для настоящего гурмана

Пицца – это одно из наиболее популярных блюд в мире,
-

Лето – это время, когда дети, закончив учебный год, уходят
Как получить гражданство Бельгии и что оно дает?

Бельгия, расположенная в сердце Европейского союза, по праву считается одним
Осетинские пироги: вкусное и популярное блюдо с Кавказа

Когда начинать готовиться к ЕГЭ и ОГЭ 2024: полезные рекомендации

Начало нового учебного года часто становится временем повышенной тревожности как
На чем можно долететь до Мальдив? Регулярный рейс или аренда частного самолета?

Путешествие на Мальдивы — это мечта многих туристов. Острова, утопающие
Зубной имплантат: преимущества выбора при протезировании

Зубной имплантат – это современная технология, предоставляющая возможность восстановить утраченный
Яйцо шоколадное Kinder сюрприз: волшебство, которое завоевало сердца детей и взрослых

Яйцо Kinder сюрприз, безусловно, является одним из наиболее популярных шоколадных
Суши и пицца: почему они так популярны в службе доставки

Службы доставки еды становятся всё популярнее среди людей, желающих насладиться
Пептидные препараты: сущность и области применения

Пептидные препараты стали одним из важнейших направлений в современной медицине