Молекулярные механизмы нейродегенерации клеток сетчатки при ишемии
Уже в начале прошлого века один из первых лауреатов Нобелевской премии Рамон-и-Кахаль назвал сетчатку «мозгом, вынесенным на периферию». Он основывался на том, что гистологически устройство мозга и сетчатки оказалось удивительно похожим. Позже выяснилось, что не только структурно эти ткани похожи. В основе их функционирования лежат одинаковые молекулярные механизмы, химические посредники, способы регуляции синаптической передачи. В отличие от других сенсорных модальностей, в процессе зрительного восприятия обработка информации происходит уже в сетчатке глаза. У многих животных такие важнейшие функции, как определение направления и скорости движения объекта, полностью локализованы в сетчатке. Известно, что чем проще устроен мозг животного, тем сложнее устроена его сетчатка.
Такое сходство приводит к тому, что и многие молекулярные механизмы, связанные с гибелью нейронов, имеют одинаковый патогенез. В ряде случаев даже при отсутствии клинических признаков заболевания мозга в сетчатке наблюдаются одинаковые молекулярные проявления. Так, при болезни Альцгеймера, причиной (не исключено что и следствием) которой является накопление β-амилоида, β-амилоид обнаруживается и в сетчатке глаза.
Другим примером является рассеянный склероз. При этом заболевании, которое характеризуется, в частности, и нарушениями функции зрительного нерва, клинические проявления неврита зрительного нерва часто являются первым эпизодом заболевания. Причиной этого является глубокая морфологическая и функциональная связь мозга и сетчатки, когда аксоны ганглиозных клеток сетчатки и формируют зрительный нерв.
Среди заболеваний, патогенез и молекулярные механизмы которых в мозге и сетчатке сходны, особое место занимает ишемия. Термин «ишемия» означает снижение кровоснабжения ткани. Для ишемии всегда характерно состояние гипоксии (недостаток кислорода) или аноксии (полное его отсутствие). Гипоксия может наблюдаться при снижении доступности кислорода или способности клеток его утилизировать, например, при снижении рО2, снижении уровня гемоглобина, повреждении митохондриальных ферментов и т.д. При ишемии кроме недостаточного снабжения ткани кислородом наблюдается дефицит питательных веществ и накопление продуктов обмена. Ишемия всегда включает понятие гипоксии, однако при гипоксии далеко не всегда наблюдаются клинические признаки ишемии.
Целью настоящей работы явилась, с одной стороны, попытка проанализировать современные представления о молекулярных механизмах развития ишемии сетчатки и выявить их сходство с такими же процессами в мозге, с другой стороны — представить полученные нами данные о ключевой роли оксида азота как необходимого фактора в процессе развития ишемии в сетчатке.
Ретинальная ишемия сопровождает большинство офтальмологических заболеваний. Ишемия сетчатки и зрительного нерва вовлечена в патогенез таких заболеваний, как окклюзии, или тромбоз ретинальных сосудов, возрастная макулярная дегенерация (ВМД), диабетическая ретинопатия (ДР), ретинопатия недоношенных (PH), глаукома. Однако, несмотря на многочисленные клинические и экспериментальные исследования, роль ретинальной ишемии в патогенезе этих заболеваний не до конца ясна. Ретинальная ишемия — клиническое состояние, при котором нарушается кровоснабжение сетчатки. Сетчатка является продолжением ЦНС и имеет много общих с мозгом эмбриологических, функциональных и анатомических признаков. Ответ на ишемию нейронов сетчатки сходен с ответом нейронов других частей ЦНС. Однако сетчатка более устойчива к ишемическому повреждению по сравнению с мозгом. Отсутствие эффективного лечения является основной причиной нарушения зрения и слепоты.
Среди молекулярных механизмов, лежащих в основе патогенеза ишемии -окислительный стресс, эксайтотоксичность, аутоиммунные и воспалительные реакции. Эти механизмы часто выступают единым фронтом, в той или иной степени проявляясь при различных заболеваниях. С другой стороны, при патологических состояниях, в развитии которых определяющую роль играет ретинальная ишемия, в спектре ее молекулярных механизмов всегда присутствуют действующие в тандеме другие ключевые механизмы ретинальных дегенераций. Например, ишемия может вызывать метаболическую дисфункцию нейронов сетчатки, стимулировать эксайтотоксичность, окислительный стресс, а индуктором окислительного стресса может выступать эксайтотоксичность.
Сетчатка особенно чувствительна к патологическим состояниям, которые нарушают сбалансированное взаимодействие ее нейрональной и сосудистой сети, обеспечивающей жизнедеятельность ретинальных нейронов. Поэтому заболевания, первично поражающие нейральную сетчатку (например дегенерация фоторецепторов), сопровождаются аномалиями сети ретинальных сосудов. С другой стороны, патологические состояния, клинически характеризуемые альтерацией хориоидальной или ретинальной сосудистой сети, такие как ВМД, ДР и PH, поражают также нейроны сетчатки, то есть всегда связаны с широким спектром гипоксических ишемических нарушений нейральной ткани. Метаболическая дисфункция нейронов при ишемии приводит к необратимым повреждениям ткани и сопровождается гибелью клеток вследствие апоптоза.
При рассмотрении молекулярных механизмов ишемических повреждений следует различать две стадии процесса — ишемию и реперфузию. Ишемия вызывается непосредственно понижением концентрации кислорода в клетках; реперфузия определяет ответ клеток на первичные ишемические изменения и возникновение компенсаторных механизмов. Основными молекулярными механизмами, определяющими патологические изменения при ишемии-реперфузии, являются энергодефицит, изменения концентрации кальция, эксайтотоксичность, окислительный стресс. Все эти процессы, как в сетчатке в целом, так и в отдельных клетках, взаимосвязаны.
К ишемии восприимчивы все нейроны сетчатки, однако наиболее чувствительными считаются ганглиозные клетки (ГК).
Фоторецепторы — специализированные клетки, имеющие самую высокую потребность в кислороде из всех клеток тела. Однако фоторецепторы менее чувствительны к ишемии, чем ГК, и для большинства ассоциированных с ишемией заболеваний наиболее характерным признаком поражения нейральной сетчатки является гибель ганглиозных клеток и истончение слоя нервных волокон. Интересно отметить, что именно уникальные для сетчатки, в отличие от мозга, структуры, а именно — фоторецепторы, наименее чувствительны к ишемическим повреждениям. Одна из причин подобных различий состоит в разной системе кровоснабжения наружных и внутренних клеточных слоев сетчатки. Кровоснабжение фоторецепторов осуществляется со стороны хориоидеи, и, поскольку фоторецепторные клетки характеризуются большой длиной, возникает градиент рО2 между базальным и апикальным полюсами клеток. Высокая плотность митохондрий во внутреннем сегменте в норме поддерживает относительно низкое рО2. Это свидетельствует о том, что фоторецепторы, вероятно, используют некие компенсаторные механизмы для выживания при низком содержании кислорода в окружающей их микросреде. На чувствительность фоторецепторов к ишемии влияет уровень световой адаптации. Светоадаптированные рецепторы менее чувствительны к ишемии и эксайтотоксичности. Палочки более чувствительны к ишемии, чем колбочки, возможно, поскольку колбочки обладают более высокой кальциевой буферной емкостью. Амакриновые клетки во внутренней сетчатке также чувствительны к ишемии и эксайтотоксичности. Ишемия повышает экспрессию провоспалительных медиаторов, таких как СОХ-2, TNF, NO, в амакриновых клетках. Эти молекулы вовлечены также в ишемическое повреждение ГК, что позволяет предполагать существование общих механизмов ишемического повреждения в различных классах нейронов. Ишемия дифференцированно поражает тела и аксоны нейронов. Это различие особенно важно для понимания патофизиологии глаукомы, где ключевые события могут развиваться в аксонах ГК скорее, чем в их клеточных телах.
При ишемии блокируется снабжение сетчатки кислородом и глюкозой, что нарушает энергетический метаболизм, приводя к снижению внутриклеточного уровня АТФ. Это, в свою очередь, нарушает мембранный потенциал и ионный гомеостаз, запуская процессы, ведущие к гибели клеток. Одним из результатов снижения энергетического метаболизма является снижение активности Na+/K+ АТФ-азы, нарушающее регуляцию мембранного потенциала.
Одним из проявлений снижения концентрации кислорода и соответствующих изменений энергообеспечения клетки при ишемии является ацидоз — закисление среды как вне клетки, так и внутри нее. Ацидоз играет важную роль в процессе ишемического повреждения ткани.
Молекулярные механизмы ацидоза идентифицированы, в основном, в экспериментах на мозге, однако предполагается, что эти механизмы являются общими для всех нервных тканей и, в частности, характерны для нейральной сетчатки. Закисление объясняется накоплением молочной кислоты, которое в условиях недостаточного снабжения кислородом вызвано переключением метаболизма с окислительного фосфорилирования на анаэробный гликолиз. Эффективность гликолитической генерации АТФ гораздо ниже аэробной, митохондриальной, поэтому процессы гидролиза АТФ начинают преобладать над синтезом этой молекулы, что способствует накоплению в клетке протонов. Интенсивный вывод протонов и лактата из клеток вызывает снижение pH вне клетки. Например, было показано, что через 10 минут тотальной ишемии мозга крысы внутриклеточный pH падал до 6,3, а содержание лактата увеличивалось в 10 раз.
Обсуждаются разные механизмы, объясняющие повреждающий эффект ацидоза при ишемии. Было показано, что при ацидозе ингибируется депонирование внутриклеточного кальция, что приводит к повышению концентрации Ca2+ в цитозоле. Повышение концентрации внутриклеточного кальция происходит в том числе и в результате активации кислото-чувствительных ионных каналов (ASICs). В норме в нервной ткани эти каналы, видимо, связаны с функционированием синапсов, вовлечены в процессы синаптической пластичности, обучения и памяти. Многочисленные эксперименты на нокаутных мышах и клеточных культурах с использованием блокаторов ASICs свидетельствуют о важной роли этих каналов в процессе ишемического повреждения нервной ткани. Еще одной причиной повреждения нейронов при ацидозе может быть усиление продукции свободных радикалов в клетке при низких значениях pH с вовлечением в этот процесс ионов железа.
Большое количество экспериментальных данных свидетельствует о том, что избыток глутамата (явление эксайтотоксичности) приводит к гибели нервных клеток и является одним из основных проявлений ишемии. Эксайтотоксичность наблюдается при таких патологиях, как ДР, глаукома, окклюзия хориоидальных и ретинальных сосудов.
Гипотезу эксайтотоксической гибели нейронов впервые выдвинул Дж.В.Е. Олней. Глутамат — основной возбуждающий нейротрансмиттер в сетчатке. Он высвобождается фоторецепторами, биполярными клетками и ГК. В норме освобожденный глутамат находится в синаптической щели очень короткое время (несколько миллисекунд), и концентрация его низка, поскольку нейроны и глия эффективно удаляют нейромедиаторы из синаптической щели после их выделения. Если уровень глутамата остается повышенным достаточно долго, это может привести возбужденные нейроны к гибели. При физиологических условиях глутамат поглощается глиальными клетками, преобразуется глутаминсинтазой в глутамин, который затем транспортируется назад к телам нейронов и там при участии глутаминазы превращается в глутамат. Снижение внутриклеточной концентрации АТФ в глиальных клетках при ишемии приводит к инактивации глутаминсинтазы, которая в таких условиях может дополнительно инактивироваться вследствие повышения уровня свободных радикалов. Метаболический баланс глутамата изменяется, и в результате сильно увеличивается соотношение глутамат/глутамин в глиальных клетках, что также может способствовать ускоренному выходу глутамата. Основная нагрузка по удалению избытка глутамата во внутренних слоях сетчатки приходится на клетки Мюллеровой глии. При ретинальной ишемии эффективность захвата глутамата этими клетками снижается. Дисфункция переносчиков глутамата в клетках глии зачастую является ключевым событием каскада реакций, ведущих к эксайтотоксической гибели нейронов сетчатки. Кроме того, снижению захвата глутамата клетками глии способствует ацидоз, развивающийся при ишемии, и генерируемые в нейронах активные формы кислорода.
Основной причиной токсического действия глутамата является увеличение внутриклеточной концентрации кальция. Активация АМРА, каинатных и NMDA-peцепторов приводит к притоку Na+ в нейроны. Это, в свою очередь, усиливает деполяризацию, которая, среди других событий, активирует потенциал-зависимые Ca2+-каналы, увеличивая входной ток Ca2+. В исследованиях на мозге было показано, что внеклеточная концентрация глутамата при ишемии повышается в два этапа: сначала наблюдается непродолжительное Са2+-зависимое повышение, за которым следует длительное Са2+-независимое повышение. На ранней стадии ишемии высвобождение глутамата увеличивается за счет активации потенциал-зависимых Са2+-каналов вследствие деполяризации мембраны, вызванной ишемией. Однако когда дефицит энергии достаточно длителен и внутриклеточный уровень АТФ становится слишком низким, чтобы обеспечивать высвобождение медиатора путем экзоцитоза, высвобождение глутамата становится Са2+-независимым в результате активации альтернативных механизмов его высвобождения.
Специфическая восприимчивость к глутамату нейронов внутренних слоев сетчатки и ганглиозных клеток при ишемии является следствием того, что эти клетки содержат ионотропные глутаматные рецепторы в высокой концентрации. Глутамат активирует эти рецепторы, вызывая избыточную деполяризацию и, в конечном счете, гибель клетки. Напротив, вызывающие гиперполяризацию тормозные медиаторы типа ГАМК ослабляют влияние глутамата, противодействуя деполяризации. Таким образом, повышение концентрации медиаторов, которые стимулируют тормозные рецепторы, защищает нейроны, содержащие эти рецепторы.
Глутамат также способствует образованию активных форм кислорода. На первых стадиях ишемии они возникают в результате нарушения кровоснабжения, а следовательно и снабжения кислородом и метаболическими субстратами. В дальнейшем, на стадии восстановления кровотока (реперфузии) и ожидаемого улучшения состояния ткани, патологический процесс продолжается вследствие так называемых реперфузионных повреждений, в которых участвуют свободные радикалы, образующиеся при повторном окислении накопленных при ишемии продуктов. Предполагают, что при значительном и достаточно быстром образовании свободных радикалов происходит подавление нормальных клеточных антиоксидантных защитных механизмов, вызывающее окислительный стресс и различные типы повреждения тканей.
Одним из участников повреждений при реперфузии является фермент ксантиноксидаза, который участвует в катаболизме пуринов, катализируя окисление гипоксантина в ксантин и его последующее окисление в мочевую кислоту. При этом в сопряженной реакции ксантиноксидаза восстанавливает кислород до супероксида, который спонтанно дисмутирует в пероксид водорода. Таким образом, в процессе этой реакции образуются активные формы кислорода, способные вызывать окислительный стресс. Так, при длительной ишемии в тканях накапливаются продукты распада пуринов, и при реперфузии, когда уровень кислорода восстанавливается, ксантиноксидаза продуцирует повышенное количество токсичных супероксида и пероксида водорода. В результате взаимодействия пероксида водорода и супероксида образуется очень токсичный гидроксильный радикал (ОН-). Кроме того, O2- может взаимодействовать с оксидом азота (NO), который образуется в значительных количествах после ишемии, приводя к формированию пероксинитрита, нитрозильного радикала и, в конечном счете, ОН-. По крайней мере, некоторые амакриновые клетки сетчатки являются дофаминергическими, и генерация активных форм кислорода во время окисления дофамина моноаминоксидазой в нервных окончаниях стимулирует развитие окислительного стресса.
Как известно, образование свободнорадикальных форм кислорода приводит к значительным повреждениям клеток нервной ткани в результате пероксидного окисления ненасыщенных жирных кислот клеточных мембран. При этом происходит как повреждение самих мембран (снижение их вязкости), так и образование вторичных пероксидных радикалов, которые могут индуцировать развитие ишемических и постишемических (при реперфузии) повреждений.
Введение в глаз взрослой крысы NMDA вызывает дозозависимую потерю ГК и холинергических амакриновых клеток, подобную той, которая наблюдается при ишемии. С другой стороны, ишемическое повреждение сетчатки может быть обратимым при добавлении антагонистов NMDA и AMPA или каината. До настоящего времени остается неясным, как взаимосвязаны процессы эксайтотоксичности и апоптотической гибели клеток при ишемии. Ишемия непосредственно запускает эксайтотоксическую нейродегенерацию, и апоптотическая нейродегенерация может развиваться как ее последствие, поскольку оставшиеся нейроны (выжившие после эксайтотоксической дегенерации) лишены синаптических контактов, необходимых для их выживания.
Ранее полагали, что ишемическая гибель клеток происходит исключительно путем классического некроза, поскольку его морфологическим критерием является набухание клеток. Гибель клеток внутренних слоев сетчатки при окклюзии центральной артерии сетчатки происходит и по пути апоптоза, и по пути некроза. Ретинальная ишемия, созданная повышением внутриглазного давления (ВГД), приводила к апоптотической гибели клеток, что было продемонстрировано гистологически, по фрагментации ДНК и в экспериментах с использованием ингибитора эндонуклеазы.
Известно, что ишемия сетчатки приводит к экспрессии каспазы-3, Вах, Bc1-x, NF-kB, разных изоформ NO-синтазы, и это лишь небольшая часть белков, участвующих в индукции апоптоза. Инактивация этих белков может предотвращать клеточную гибель при ретинальной ишемии, как, например, это было показано в экспериментах с использованием специфических ингибиторов каспаз. Это позволяет надеяться, что создание фармакологических средств, предотвращающих развитие апоптоза, может предотвратить дегенерацию клеток сетчатки при заболеваниях глаза, обусловленных ишемией.
Что касается апоптотической гибели клеток с участием NO-синтазы (NOS), а следовательно и NO, то повышение экспрессии нейрональной и индуцибельной NO-синтазы (nNOS и iNOS, соответственно) при ретинальной ишемии продемонстрировано на различных экспериментальных моделях. При ишемии повышается внутриклеточная концентрация кальция, что приводит к активации NOS и повышению концентрации NO.
Предполагается, что нейротоксичность NO обусловлена образованием пероксинитрита (ONOO-) при взаимодействии NO с супероксид-анион-радикалом. In vitro скорость этой реакции в три раза выше, чем активность супероксиддисмутазы (СОД). Таким образом, при значительных концентрациях NO эффективно конкурирует с СОД за супероксид-анион.
Повышенное образование супероксидного анион-радикала при ишемии наблюдается на стадии реперфузии. Химическое действие пероксинитрита достаточно сложно. Он может проявлять активность и как гидроксильный радикал, и как радикал двуокиси азота. Пероксинитрит может нитровать и гидроксилировать ароматические кольца аминокислотных остатков, а также окислять ДНК и сульфгидрильные группы белков, что может являться сигналом для апоптоза.
Для изучения молекулярных механизмов развития ишемии нервных тканей в сетчатке принципиальное значение имеет выбор соответствующей модели. Ишемия мозга в эксперименте достигается обычно путем кратковременной окклюзии сонной артерии, что позволяет изучать как механизм ишемии, так и механизм реперфузии. Существует несколько методик создания экспериментальной ишемии, наиболее распространенными из которых являются искусственное повышение внутриглазного давления или наложение лигатуры на сосуды в области зрительного нерва. Каждый из этих подходов имеет свои недостатки. Метод повышения внутриглазного давления очень чувствителен к любым механическим воздействиям, что делает результаты нестабильными. Кроме того, повышение ВГД неизбежно ведет к дегенерации ганглиозных клеток, которая может и не быть обусловлена ишемией. Во втором случае при наложении лигатуры неизбежно повреждаются не только сосуды, но и зрительный нерв, что приводит к независимым от ишемии дополнительным патологическим изменениям в сетчатке. Таким образом, имеющиеся модели не могут дифференцировать гибель клеток сетчатки, вызванную ишемией, от гибели клеток, обусловленной независимой от ишемии дегенерацией клеток сетчатки. Понимание отличий этих механизмов патогенеза принципиально важно как для поиска перспективных лекарственных средств, так и для выбора правильной стратегии лечения. Так, антиишемические средства могут и не предотвращать гибель нейронов, вызванную другими причинами.
Избежать упомянутых недостатков при моделировании ишемии можно путем непосредственного локального воздействия лазерного излучения на магистральные ретинальные сосуды, что вполне выполнимо, поскольку они хорошо видны и доступны для лазерной коагуляции. Эта модель является удобной, так как позволяет избежать, с одной стороны, указанных выше недостатков, а с другой — получать избирательное тромбирование сосудов, создаваемое лазерной коагуляцией. Это позволяет, выбирая различные отделы магистральных ретинальных сосудов или их ветвей и изменяя параметры лазерного излучения, создавать как локальную, так и обширную ишемию сетчатки. Предлагаемая нами модель с использованием лазерной коагуляции имеет ряд преимуществ. Во-первых, изменение скорости кровотока происходит не в результате длительного (1-2 часа) воздействия, а в доли секунды (время экспозиции лазерного излучения). Это позволяет изучать ишемические нарушения в остром периоде. Во-вторых, модель быстро и легко реализуема при наличии лазерного оборудования и, в-третьих, лазерная коагуляция моделирует такое офтальмологическое заболевание, как тромбоз ретинальных сосудов. На описанной выше модели нами продемонстрировано также защитное действие оксида азота от ишемических повреждений клеток сетчатки. При этом важными являются два аспекта. Во-первых, ретинальные сосуды хорошо видны в офтальмоскопе: действие сосудорасширяющих факторов можно наблюдать в данном случае в реальном времени. По существу, такой подход позволяет визуально наблюдать за реакцией сосуда на гипоксию in vivo. Поскольку мы предполагали, что нитриты будут восстанавливаться в сосуде при гипоксии, это должно было приводить к резкому расширению сосуда. Во-вторых, коагуляция сосуда вызывает ишемию сетчатки и, если нитриты действительно восстанавливаются в сосуде при гипоксии, это должно было косвенно проявиться как защитный эффект при развитии ишемии сетчатки. Наиболее удобной для этих исследований оказалась модель ишемии на кроликах. Ретинальные сосуды кролика хорошо видны, что позволяет легко наблюдать их реакцию на лазерное воздействие. Кроме того, моделирование ишемии на кроликах позволит в перспективе использовать метод ОСТ (ocular computer tomography) для оценки тонких изменений сетчатки при ишемии, а также реконструировать трехмерное изображение ретинальных сосудов и точно измерять их диаметр.
Лазерная коагуляция ретинальных сосудов приводит к значительным изменениям в кровенаполнении сосудов. Если в норме сосуды сетчатки в области диска зрительного нерва не извиты и соответствуют средней норме, то сразу после лазерного воздействия видно, что сосуды запустели и резко уменьшились в диаметре (рис. 1 а,б).
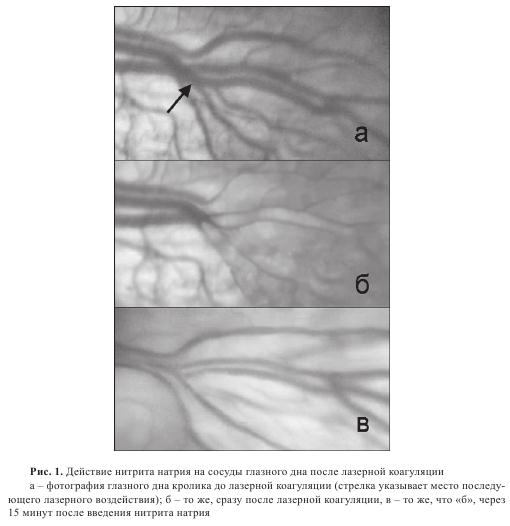
Для выяснения возможного действия нитритов на сетчатку глаза и ретинальные сосуды нитрит натрия вводили животным как до, так и после проведения лазерной коагуляции. В том случае, когда нитрит натрия вводили кролику в течение 15 минут после лазерной коагуляции в количестве 20 мг/кг массы, наблюдалось восстановление кровенаполнения сосудов непосредственно за зоной лазерного воздействия (рис. 1в). В самих поврежденных сосудах начинается восстановление кровообращения. Таким образом, введение нитрита натрия на фоне гипоксии приводит к быстрому расслаблению сосудов, что свидетельствует о том, что в условиях ишемии нитрит быстро восстанавливается до оксида азота. При предварительном введении нитритов картина была другой. Животному сначала вводили нитрит натрия (20 мг/ кг), а затем, через 15 минут, производили лазерную коагуляцию и наблюдали за изменением сосудов глазного дна (рис. 2). В этом случае лазерное воздействие создавало не стойкий, а лишь кратковременный спазм сосуда с быстрым последующим восстановлением кровенаполнения и проводимости сосуда.
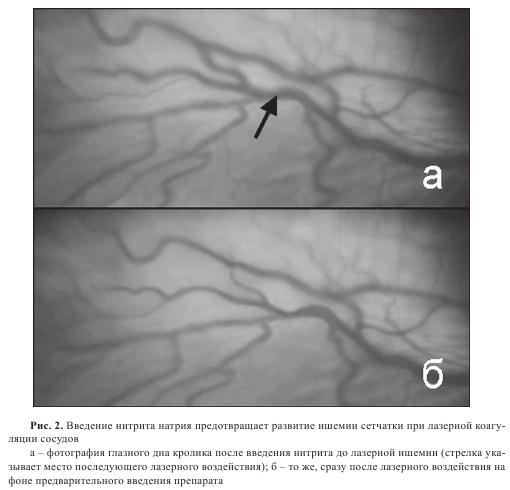
Таким образом, можно заключить, что нитрит натрия в условиях острой гипоксии приводит к почти мгновенному расширению сосудов, что и делает лазерную коагуляцию неэффективной. Можно предполагать, что в условиях гипоксии, когда нитриты могут связываться с гемом гемсодержащих белков, может происходить их восстановление до оксида азота в концентрациях, достаточных для быстрого и полного расслабления сосудов.
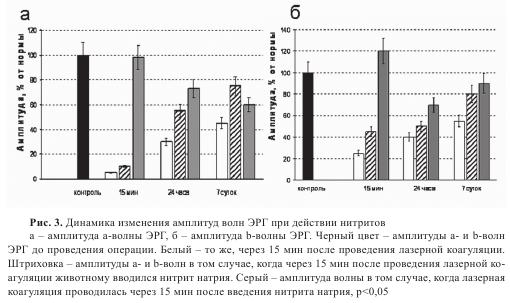
Известно, что ишемия сетчатки сопровождается характерными изменениями в картине фотоэлектрической активности сетчатки — электроретинограмме (ЭРГ). Использование ЭРГ позволило оценить действие нитритов на ретинальные сосуды по изменению электрической реакции сетчатки глаза, то есть независимым методом. Регистрация ЭРГ выполнялась сразу после лазерной коагуляции, после введения препарата на фоне лазерного воздействия и после лазерного воздействия на фоне предварительного введения нитрита натрия. Лазерная коагуляция сосудов сетчатки приводила к резкому снижению амплитуд а- и b-волн ЭРГ (рис. 3). Введение нитрита натрия в дозе 20 мг/кг массы через 15 минут после лазерной коагуляции приводило к более быстрому восстановлению а- и b-волн ЭРГ. Наибольший положительный эффект препарата был выявлен при его введении до создания лазерной ишемии сетчатки, где не наблюдалось угнетения биопотенциалов.
Глиальный индекс Kг, рассчитываемый по отношению амплитуд b-волны ЭРГ и РЭРГ (12 Гц), равнялся 1,8 при норме 2,3-2,5 относительных единиц. Известно, что возрастание глиального индекса, отражающее активизацию метаболизма клеток Мюллера, является характерным признаком ретинальной ишемии. Однако при острых нарушениях кровообращения в бассейне центральной артерии сетчатки b-волна ЭРГ быстрее реагирует на гипоксию сетчатки, связанную с сосудистой катастрофой, чем низкочастотная РЭРГ. Поэтому опережающее снижение амплитуды b-волны ЭРГ сразу после лазерной коагуляции сосудов приводит к снижению Kг, отражая значительные нарушения в сетчатке. Действительно, клинические наблюдения свидетельствуют о развитии обтурации, кровотечения и запустения сосудов после лазерного воздействия и последующей острой ишемии сетчатки.
Таким образом, полученные нами двумя независимыми методами (ЭРГ и офтальмоскопия) результаты показывают, что введение нитритов экспериментальным животным защищает сетчатку от острой ишемии. Такое действие, по-видимому, обусловлено тем, что при недостатке кислорода in vivo нитрит натрия может восстанавливаться до оксида азота в концентрациях, достаточных для полного расслабления сосудов.
Модель ишемии сетчатки глаза на кроликах в некоторых экспериментах очень удобна, т.к. сосуды глаза хорошо видны, и кровоснабжение сетчатки кролика отличается от кровоснабжения сетчатки человека. У кроликов 2/3 сетчатки питается от хориоидальных сосудов, которые снабжают наружные слои сетчатки. И всего 1/3 сетчатки питается от ретинальных сосудов, которые распадаются на капилляры в пределах ганглиозного слоя и слоя нервных волокон. В то же время у человека, наоборот, 1/3 кровоснабжения осуществляется через хориоидальные сосуды и 2/3 — через ретинальные. Важно отметить, что основной причиной зрительных патологий человека является ишемия, вызванная нарушениями именно в ретинальных сосудах. Наиболее близким объектом для моделирования заднего отрезка глаза является крыса, у которой кровоснабжение сетчатки осуществляется так же, как у человека.
При офтальмоскопии глазного дна сразу после лазерной коагуляции отмечалось резкое сужение магистральных сосудов с полной или почти полной остановкой кровотока, сохраняющейся на протяжении нескольких минут, после чего кровоток частично восстанавливался, но просвет сосуда оставался значительно суженным. На следующий день после операции у некоторых животных наблюдалась стушеванность границ диска зрительного нерва из-за отека окружающей сетчатки, суженные артерии, местами до полной их непроходимости, кровоток в них становился прерывистым. Некоторое расширение вен, по-видимому, было обусловлено одновременным нарушением венозной гемодинамики в результате распространения коагулирующего действия лазерного излучения на стенку расположенного рядом магистрального венозного сосуда.
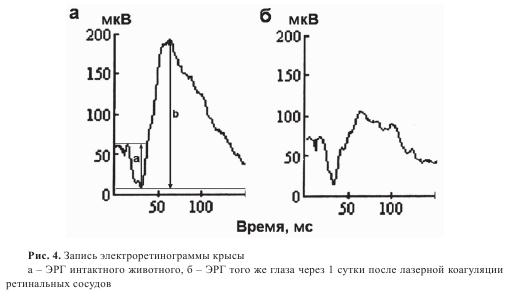
Через сутки после операции в результате тромбоза магистральных ретинальных сосудов отмечались достаточно выраженные гемодинамические нарушения в ретинальной системе микроциркуляции, приводящие к ишемии внутренних слоев сетчатки. Стойкие гемодинамические нарушения в системе ретинального кровотока через сутки приводили к электрофизиологическим изменениям в сетчатке, характерным для ишемии. Для оценки степени развития ишемии сетчатки широко используется соотношение амплитуд b- и a-волн ЭРГ — индекс b/а. а-волна ЭРГ характеризует функциональную активность фоторецепторных клеток, b-волна — внутренних слоев сетчатки, прежде всего биполярных клеток. На следующий день после лазерной коагуляции сосудов амплитуда b-волны ЭРГ снижалась на 40-70% от исходных значений (рис. 4). Угнетение a-волны ЭРГ отмечалось через сутки не у всех животных. В тех случаях, когда амплитуда а-волны снижалась, угнетение b-волны было более значительным. Изменения b-волны развиваются, как правило, раньше угнетения a-волны и являются более выраженными из-за ухудшения трофики нейронов внутреннего ядерного слоя сетчатки при нарушениях ретинальной циркуляции. В нашем исследовании при развитии острой ишемии сетчатки коэффициент b/а резко снижался — до 1,0-2,0 отн. единиц при его нормальных значениях у крыс 3,3-4,8 ед.
Данные электроретинографических исследований подтверждаются гистологическими результатами. Нарушение кровотока в ретинальных сосудах вследствие их тромбирования уже через сутки приводило к ишемическим повреждениям во внутренних слоях сетчатки глаза (рис. 5). Типичными проявлениями ишемического повреждения были кровоизлияния, микрокистовидные изменения в слое нервных волокон, отек ганглиозных клеток, локальное уменьшение плотности нейронов в слое биполярных клеток. Следствием периваскулярного отека явилось истончение и микроразрывы внутренней пограничной мембраны, через которые происходила миграция мононуклеарных клеток крови из просвета сосуда в корковые отделы прилежащего стекловидного тела.

Таким образом, результаты наших электрофизиологических и гистологических исследований позволяют заключить, что лазерная коагуляция ретинальных сосудов приводит к развитию характерных ишемических изменений в сетчатке.
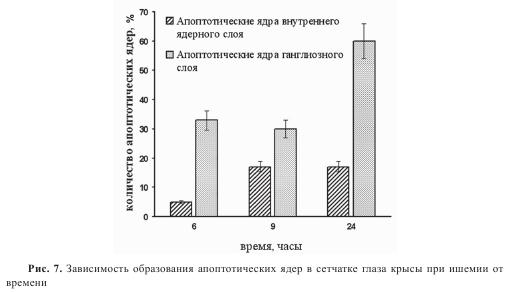
Для подтверждения того, что гибель клеток идет по апоптотическому пути, был применен метод TUNEL. Данным методом были выявлены TUNEL-положительные ядра в ганглиозном слое уже через 6 часов после лазерного воздействия на сосуды (рис. 6). Также наблюдался апоптоз амакриновых и ганглиозных клеток через 18 и 24 часа после лазерной коагуляции. При лазерной коагуляции ретинальных сосудов наибольшее количество апоптотических ядер в ганглиозном слое было выявлено через 24 часа после тромбирования сосудов (60% от общего числа клеток) (рис. 7). Максимальное количество TUNEL-положительных ядер во внутреннем ядерном слое (ВЯС) было обнаружено уже через 9 часов после лазерного воздействия.
Понимание молекулярного механизма гибели клеток при патологии особенно важно при попытках фармакологически предотвратить их гибель. Известно, что гибель нервных клеток при развитии ишемии может идти апоптотическим путем.
Можно предполагать, что поскольку повышенная концентрация нитритов в сосудах защищала сетчатку от появления признаков ишемии, то этот механизм должен предотвращать и развитие апоптоза клеток сетчатки.
Иммуногистохимическим методом TUNEL было показано, что предварительное введение нитрита натрия животным приводило к значительному снижению количества TUNEL-положительных ядер в ганглиозном слое и защищало от апоптоза клетки внутреннего ядерного слоя (рис. 8). При предварительном введении нитрита натрия до лазерной коагуляции сосудов сетчатки количество апоптотических клеток в ганглиозном слое сетчатки снижалось на 66%, а во внутреннем ядерном слое — на 50% (рис. 9). Важно отметить, что изменения затрагивали, главным образом, внутренние отделы сетчатки (до ВЯС), которые кровоснабжаются ретинальными сосудами. В наружных отделах, которые питаются из слоя хориокапилляров (слой наружных сегментов фоторецепторов, наружный ядерный слой), видимых морфологических изменений не обнаружено.

Известно, что гибель нервных клеток мозга и сетчатки при некоторых патологиях происходит апоптотическим путем при участии оксида азота. Естественно предполагать, что и при развитии ишемии оксид азота может быть индуктором развития апоптоза. Ранее было установлено, что введение животным конкурентного ингибитора NO-синтазы L-NAME приводит к существенному снижению концентрации оксида азота в клеточных слоях сетчатки и поэтому можно предполагать, что введение этого ингибитора будет влиять на развитие апоптоза. Для подтверждения этого экспериментальным животным вводили ингибитор NO-синтазы L-NAME. Внутрибрюшинное введение этого препарата в дозе 20 мг/кг веса снижало концентрацию оксида азота в клетках сетчатки, и пероксинитрит, очевидно, не образовывался. При предварительном введении L-NAME за 15 минут до лазерного воздействия на ретинальные сосуды апоптотические клетки во внутренних слоях сетчатки не выявлялись (рис. 10). Лазерная коагуляция сосудов сетчатки глаза приводит к гибели 50% клеток ганглиозного слоя и 30% клеток ВЯС сетчатки. При предварительном введении препарата L-NAME крысам лазерное воздействие на ретинальные сосуды приводит к гибели всего 20% ганглиозных клеток и 15% клеток ВЯС от общего числа клеток (рис. 11). Можно сделать вывод, что ингибитор NO-синтазы L-NAME защищает клетки сетчатки от апоптоза при ишемии. Таким образом, оксид азота является необходимым элементом развития апоптоза в клеточных слоях сетчатки.

Глутаматная интоксикация может являться одним из звеньев длинной цепи ишемического повреждения, но может происходить и независимо от ишемии при различных дегенеративных нарушениях. В работе нами исследовались влияние повышения концентрации глутамата на возникновение нейродегенеративных нарушений в сетчатке и роль оксида азота в этих процессах. NMDA (агонист глутамата) вводили непосредственно в глаз экспериментальным животным в дозе 200 нмоль/глаз. И уже через сутки методом TUNEL можно было выявить апоптотические ядра во внутреннем ядерном слое и слое ганглиозных клеток сетчатки (рис. 12). Введение NMDA приводит к апоптотической гибели 60% ганглиозных клеток от общего числа клеток ганглиозного слоя и 50% клеток внутреннего ядерного слоя от общего числа клеток этого слоя. ЭРГ также ясно свидетельствует о снижении функциональной активности во внутренних слоях сетчатки: наблюдается уменьшение амплитуды b-волны на 50% от нормы (рис. 13).

Известно, что передача сенсорного сигнала от фоторецепторов к последующим слоям сетчатки осуществляется посредством глутамата. Введение агониста глутамата NMDA приводит к блокированию этой передачи и увеличению концентрации глутумата в синаптической щели. Это приводит к тому что ответы фоторецепторных клеток сохраняются, а ответы более глубоких слоев сетчатки значительно уменьшаются. Именно такая картина наблюдается и при электроретинографических исследованиях. Как видно, а-волна ЭРГ, которая обусловлена электрической активностью фоторецепторов, практически не изменяется, а b-волна, основной вклад в которую вносят ответы биполярных клеток, существенно уменьшена.
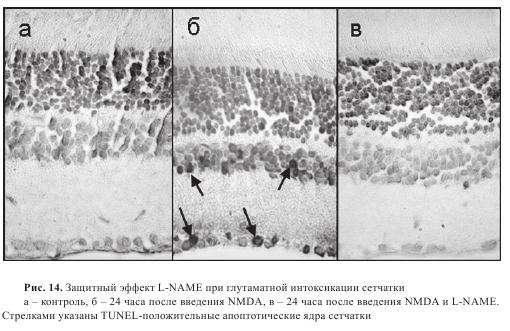
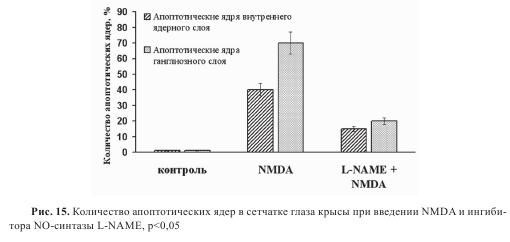
Введение NMDA приводило к развитию апоптоза в клеточных слоях сетчатки. Так, через 24 часа после введения апоптотические ядра обнаруживались во внутреннем ядерном слое и слое ганглиозных клеток, то есть именно в тех слоях, где медиаторная роль глутамата установлена. Для того чтобы выяснить, по какому пути идет гибель клеток, в стекловидное тело глаза вместе с NMDA вводили ингибитор NO-синтазы L-NAME (0,1 мг/глаз). При этом количество апоптотических ядер в сетчатке резко снижалось (рис. 14). Введение L-NAME вместе с NMDA в стекловидное тело глаза приводило к снижению количества апоптотических ядер во внутреннем ядерном слое и слое ганглиозных клеток на 50% и 70%, соответственно (рис. 15).
Для подтверждения предположения, что в разработанных нами экспериментальных моделях глазных патологий именно высокая концентрация NO является токсическим фактором, нам требовалось создать 20-кратный избыток NO в сетчатке. Для этого мы вводили в глаз донор оксида азота динитрозильный комплекс железа с глутатионом (ДНК-Ж). Динитрозильные комплексы железа в тканях животных и человека продуцируют свободные молекулы оксида азота и могут функционировать в качестве эндогенных универсальных регуляторов биохимических и физиологических процессов. ДНК-Ж обеспечивают стабилизацию и перенос NO в биосистемах. Так как компоненты этого комплекса уже содержатся в системе, то его введение не является токсичным для клеток сетчатки. Ниже приведена структурная формула ДНК-Ж с глутатионом и распад этого соединения на составляющие компоненты с образованием оксида азота.
Введение в стекловидное тело глаза крыс динитрозильного комплекса железа с глутатионом (10в-5-10в-8 моль/глаз) через сутки приводило к появлению апоптотических ядер в сетчатке. Методом TUNEL было выявлено, что апоптозу подверглись клетки внутреннего ядерного и ганглиозного слоев сетчатки (рис. 16). При этом апоптоз был выявлен только при введении ДНК-Ж в дозах 10в-7 и 10в-8 моль/глаз. А при введении большего количества препарата (10в-6 и 10в-5 моль/глаз) TUNEL-положительные ядра в сетчатке выявить не удалось. При введении ДНК-Ж в количестве 10в-7 моль/глаз количество апоптотических клеток в ганглиозном слое сетчатки увеличилось на 30% от нормы (рис. 17). При введении препарата в количестве 10в-8 моль/глаз апоптозу подверглось до 70% клеток ганглиозного слоя сетчатки от общего количества клеток. При этом клетки внутреннего ядерного слоя подвергались апоптозу в незначительной степени: их количество составило всего 15% от нормы.
Таким образом, введение донора оксида азота ДНК-Ж в низких дозах приводит к апоптозу клеток сетчатки экспериментальных животных, так как избыток NO в ткани ведет к образованию пероксинитрита, а следовательно и к гибели клеток.


Читайте также
Услуги гидроизоляции — внешняя или внутренняя?

Трудовые споры: как добиться справедливости от недобросовестного работодателя

Трудовые отношения — это тонкая материя, полная нюансов и правовых
Как отличить брендовые очки от подделки

Брендовые солнцезащитные очки — это не только модный аксессуар, но
Дизайн встроенной кухни: как оптимизировать пространство

Несмотря на большое разнообразие готовой (типовой) мебели, мебель на заказ
Михаил Владимирович Мишустин: отличный управленец и экономист

Михаил Владимирович Мишустин — выдающийся российский государственный и политический деятель,
Самые популярные рецепты пиццы: идеальное сочетание ингредиентов для настоящего гурмана

Пицца – это одно из наиболее популярных блюд в мире,
-

Лето – это время, когда дети, закончив учебный год, уходят
Как получить гражданство Бельгии и что оно дает?

Бельгия, расположенная в сердце Европейского союза, по праву считается одним
Осетинские пироги: вкусное и популярное блюдо с Кавказа

Когда начинать готовиться к ЕГЭ и ОГЭ 2024: полезные рекомендации

Начало нового учебного года часто становится временем повышенной тревожности как
На чем можно долететь до Мальдив? Регулярный рейс или аренда частного самолета?

Путешествие на Мальдивы — это мечта многих туристов. Острова, утопающие
Зубной имплантат: преимущества выбора при протезировании

Зубной имплантат – это современная технология, предоставляющая возможность восстановить утраченный
Яйцо шоколадное Kinder сюрприз: волшебство, которое завоевало сердца детей и взрослых

Яйцо Kinder сюрприз, безусловно, является одним из наиболее популярных шоколадных
Суши и пицца: почему они так популярны в службе доставки

Службы доставки еды становятся всё популярнее среди людей, желающих насладиться
Пептидные препараты: сущность и области применения

Пептидные препараты стали одним из важнейших направлений в современной медицине